Analysis of Early Evolutionary Events During the Transition from Prokaryotes to Eukaryotes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Extremozymes of the Hot and Salty Halothermothrix Orenii
Extremozymes of the Hot and Salty Halothermothrix orenii Author Kori, Lokesh D Published 2012 Thesis Type Thesis (PhD Doctorate) School School of Biomolecular and Physical Sciences DOI https://doi.org/10.25904/1912/2191 Copyright Statement The author owns the copyright in this thesis, unless stated otherwise. Downloaded from http://hdl.handle.net/10072/366220 Griffith Research Online https://research-repository.griffith.edu.au Extremozymes of the hot and salty Halothermothrix orenii LOKESH D. KORI (M.Sc. Biotechnology) School of Biomolecular and Physical Sciences Science, Environment, Engineering and Technology Griffith University, Australia Submitted in fulfillment of the requirements of the degree of Doctor of Philosophy December 2011 STATEMENT OF ORIGINALITY STATEMENT OF ORIGINALITY This work has not previously been submitted for a degree or diploma in any university. To the best of my knowledge and belief, the thesis contains no material previously published or written by another person except where due reference is made in the thesis itself. LOKESH DULICHAND KORI II ACKNOWLEDGEMENTS ACKNOWLEDGEMENTS I owe my deepest gratitude to my supervisor Prof. Bharat Patel, for offering me an opportunity for being his postgraduate. His boundless knowledge motivates me for keep going and enjoy the essence of science. Without his guidance, great patience and advice, I could not finish my PhD program successfully. I take this opportunity to give my heartiest thanks to Assoc. Prof. Andreas Hofmann, (Structural Chemistry, Eskitis Institute for Cell & Molecular Therapies, Griffith University) for his support and encouragement for crystallographic work. I am grateful to him for teaching me about the protein structures, in silico analysis and their hidden chemistry. -

The Life of Brine: Halophiles in 2001 Comment Mike Dyall-Smith* and Michael Danson
http://genomebiology.com/2001/2/12/reports/4033.1 Meeting report The life of brine: halophiles in 2001 comment Mike Dyall-Smith* and Michael Danson Addresses: *Department of Microbiology and Immunology, University of Melbourne, Victoria 3010, Australia. Centre for Extremophile Research, Department of Biology and Biochemistry, University of Bath, Bath, BA2 7AY, UK. Correspondence: Mike Dyall-Smith. E-mail: [email protected] Published: 21 November 2001 reviews Genome Biology 2001, 2(12):reports4033.1–4033.3 The electronic version of this article is the complete one and can be found online at http://genomebiology.com/2001/2/12/reports/4033 © BioMed Central Ltd (Print ISSN 1465-6906; Online ISSN 1465-6914) reports haloarchaea found in salt ponds that have 8% salt (or A report on the International conference on Halophilic 2.3-fold concentrated seawater) but are not detectable at the Microorganisms, Sevilla, Spain, 23-27 September 2001. higher salinities usually associated with haloarchaea (20-35% salt). Discovered using 16S rRNA libraries, the new haloar- chaea are distantly related to Haloarcula. None has been cul- The 2001 halophiles meeting covered archaea, bacteria, tured but these results suggest an optimum salt concentration fungi and algae adapted to living hypersaline environments. for growth far lower than that of known haloarchaea. These deposited research Despite the absence of some American colleagues as a result represent an intermediate group of haloarchaea, not previ- of the recent terrorist attack, the meeting was full of exciting ously suspected, and offer insight into the evolution of advances (as well as pictures of salt lakes, salterns, brines extreme halophiles. -

The Phagotrophic Origin of Eukaryotes and Phylogenetic Classification Of
International Journal of Systematic and Evolutionary Microbiology (2002), 52, 297–354 DOI: 10.1099/ijs.0.02058-0 The phagotrophic origin of eukaryotes and phylogenetic classification of Protozoa Department of Zoology, T. Cavalier-Smith University of Oxford, South Parks Road, Oxford OX1 3PS, UK Tel: j44 1865 281065. Fax: j44 1865 281310. e-mail: tom.cavalier-smith!zoo.ox.ac.uk Eukaryotes and archaebacteria form the clade neomura and are sisters, as shown decisively by genes fragmented only in archaebacteria and by many sequence trees. This sisterhood refutes all theories that eukaryotes originated by merging an archaebacterium and an α-proteobacterium, which also fail to account for numerous features shared specifically by eukaryotes and actinobacteria. I revise the phagotrophy theory of eukaryote origins by arguing that the essentially autogenous origins of most eukaryotic cell properties (phagotrophy, endomembrane system including peroxisomes, cytoskeleton, nucleus, mitosis and sex) partially overlapped and were synergistic with the symbiogenetic origin of mitochondria from an α-proteobacterium. These radical innovations occurred in a derivative of the neomuran common ancestor, which itself had evolved immediately prior to the divergence of eukaryotes and archaebacteria by drastic alterations to its eubacterial ancestor, an actinobacterial posibacterium able to make sterols, by replacing murein peptidoglycan by N-linked glycoproteins and a multitude of other shared neomuran novelties. The conversion of the rigid neomuran wall into a flexible surface coat and the associated origin of phagotrophy were instrumental in the evolution of the endomembrane system, cytoskeleton, nuclear organization and division and sexual life-cycles. Cilia evolved not by symbiogenesis but by autogenous specialization of the cytoskeleton. -

Diagnosis and Differentiation of the Order Primates
YEARBOOK OF PHYSICAL ANTHROPOLOGY 30:75-105 (1987) Diagnosis and Differentiation of the Order Primates FREDERICK S. SZALAY, ALFRED L. ROSENBERGER, AND MARIAN DAGOSTO Department of Anthropolog* Hunter College, City University of New York, New York, New York 10021 (F.S.S.); University of Illinois, Urbanq Illinois 61801 (A.L. R.1; School of Medicine, Johns Hopkins University/ Baltimore, h4D 21218 (M.B.) KEY WORDS Semiorders Paromomyiformes and Euprimates, Suborders Strepsirhini and Haplorhini, Semisuborder Anthropoidea, Cranioskeletal morphology, Adapidae, Omomyidae, Grades vs. monophyletic (paraphyletic or holophyletic) taxa ABSTRACT We contrast our approach to a phylogenetic diagnosis of the order Primates, and its various supraspecific taxa, with definitional proce- dures. The order, which we divide into the semiorders Paromomyiformes and Euprimates, is clearly diagnosable on the basis of well-corroborated informa- tion from the fossil record. Lists of derived features which we hypothesize to have been fixed in the first representative species of the Primates, Eupri- mates, Strepsirhini, Haplorhini, and Anthropoidea, are presented. Our clas- sification of the order includes both holophyletic and paraphyletic groups, depending on the nature of the available evidence. We discuss in detail the problematic evidence of the basicranium in Paleo- gene primates and present new evidence for the resolution of previously controversial interpretations. We renew and expand our emphasis on postcra- nial analysis of fossil and living primates to show the importance of under- standing their evolutionary morphology and subsequent to this their use for understanding taxon phylogeny. We reject the much advocated %ladograms first, phylogeny next, and scenario third” approach which maintains that biologically founded character analysis, i.e., functional-adaptive analysis and paleontology, is irrelevant to genealogy hypotheses. -

Diversity of Halophilic Archaea in Fermented Foods and Human Intestines and Their Application Han-Seung Lee1,2*
J. Microbiol. Biotechnol. (2013), 23(12), 1645–1653 http://dx.doi.org/10.4014/jmb.1308.08015 Research Article Minireview jmb Diversity of Halophilic Archaea in Fermented Foods and Human Intestines and Their Application Han-Seung Lee1,2* 1Department of Bio-Food Materials, College of Medical and Life Sciences, Silla University, Busan 617-736, Republic of Korea 2Research Center for Extremophiles, Silla University, Busan 617-736, Republic of Korea Received: August 8, 2013 Revised: September 6, 2013 Archaea are prokaryotic organisms distinct from bacteria in the structural and molecular Accepted: September 9, 2013 biological sense, and these microorganisms are known to thrive mostly at extreme environments. In particular, most studies on halophilic archaea have been focused on environmental and ecological researches. However, new species of halophilic archaea are First published online being isolated and identified from high salt-fermented foods consumed by humans, and it has September 10, 2013 been found that various types of halophilic archaea exist in food products by culture- *Corresponding author independent molecular biological methods. In addition, even if the numbers are not quite Phone: +82-51-999-6308; high, DNAs of various halophilic archaea are being detected in human intestines and much Fax: +82-51-999-5458; interest is given to their possible roles. This review aims to summarize the types and E-mail: [email protected] characteristics of halophilic archaea reported to be present in foods and human intestines and pISSN 1017-7825, eISSN 1738-8872 to discuss their application as well. Copyright© 2013 by The Korean Society for Microbiology Keywords: Halophilic archaea, fermented foods, microbiome, human intestine, Halorubrum and Biotechnology Introduction Depending on the optimal salt concentration needed for the growth of strains, halophilic microorganisms can be Archaea refer to prokaryotes that used to be categorized classified as halotolerant (~0.3 M), halophilic (0.2~2.0 M), as archaeabacteria, a type of bacteria, in the past. -
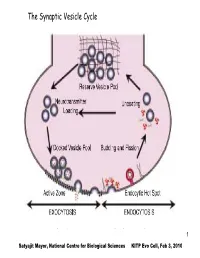
The Synaptic Vesicle Cycle
The Synaptic Vesicle Cycle 1 Satyajit Mayor, National Centre for Biological Sciences KITP Evo Cell, Feb 3, 2010 The Eukaryotic Commandments: T. Cavalier-Smith (i)origin of the endomembrane system (ER, Golgi and lysosomes) and coated- vesicle budding and fusion, including endocytosis and exocytosis; (ii) origin of the cytoskeleton, centrioles, cilia and associated molecular motors; (iii) origin of the nucleus, nuclear pore complex and trans-envelope protein and RNA transport; (iv) origin of linear chromosomes with plural replicons, centromeres and telomeres; (v) origin of novel cell-cycle controls and mitotic segregation; (vi) origin of sex (syngamy, nuclear fusion and meiosis); (vii) origin of peroxisomes; (ix) origin of mitochondria; (viii) novel patterns of rRNAprocessing using small nucleolar-ribonucleoproteins (snoRNPs); (x) origin of spliceosomal introns. 2 EM of Chemical synapse mitochondria Active Zone Figure 5.3, Bear, 2001 3 T. Cavalier-Smith International Journal of Systematic and Evolutionary Microbiology (2002), 52, 297–354 Fig. 1. The bacterial origins of eukaryotes as a two-stage process. I phase: The ancestors of eukaryotes, the stem neomura, are shared with archaebacteria and evolved during the neomuran revolution, in which N-linked glycoproteins replaced murein peptidoglycan and 18 other suites of characters changed radically through adaptation of an ancestral actinobacterium to thermophily, as discussed in detail by Cavalier- Smith (2002a). 4 T. Cavalier-Smith International Journal of Systematic and Evolutionary Microbiology (2002), 52, 297–354 Fig. 1. The bacterial origins of eukaryotes as a two-stage process. II phase: In the next phase, archaebacteria and eukaryotes diverged dramatically. Archaebacteria retained the wall and therefore their general bacterial cell and genetic organization, but became adapted to even hotter and more acidic environments by substituting prenyl ether lipids for the ancestral acyl esters and making new acid- resistant flagellar shafts (Cavalier-Smith, 2002a). -

A Revised Taxonomy of the Iguanodont Dinosaur Genera and Species
ARTICLE IN PRESS + MODEL Cretaceous Research xx (2007) 1e25 www.elsevier.com/locate/CretRes A revised taxonomy of the iguanodont dinosaur genera and species Gregory S. Paul 3109 North Calvert Station, Side Apartment, Baltimore, MD 21218-3807, USA Received 20 April 2006; accepted in revised form 27 April 2007 Abstract Criteria for designating dinosaur genera are inconsistent; some very similar species are highly split at the generic level, other anatomically disparate species are united at the same rank. Since the mid-1800s the classic genus Iguanodon has become a taxonomic grab-bag containing species spanning most of the Early Cretaceous of the northern hemisphere. Recently the genus was radically redesignated when the type was shifted from nondiagnostic English Valanginian teeth to a complete skull and skeleton of the heavily built, semi-quadrupedal I. bernissartensis from much younger Belgian sediments, even though the latter is very different in form from the gracile skeletal remains described by Mantell. Currently, iguanodont remains from Europe are usually assigned to either robust I. bernissartensis or gracile I. atherfieldensis, regardless of lo- cation or stage. A stratigraphic analysis is combined with a character census that shows the European iguanodonts are markedly more morpho- logically divergent than other dinosaur genera, and some appear phylogenetically more derived than others. Two new genera and a new species have been or are named for the gracile iguanodonts of the Wealden Supergroup; strongly bipedal Mantellisaurus atherfieldensis Paul (2006. Turning the old into the new: a separate genus for the gracile iguanodont from the Wealden of England. In: Carpenter, K. (Ed.), Horns and Beaks: Ceratopsian and Ornithopod Dinosaurs. -

Halophilic Microorganisms Springer-Verlag Berlin Heidelberg Gmbh Antonio Ventosa (Ed.)
Halophilic Microorganisms Springer-Verlag Berlin Heidelberg GmbH Antonio Ventosa (Ed.) Halophilic Microorganisms With 58 Figures Springer Dr.ANTONIO VENTOSA Department of Microbiology and Parasitology Faculty of Pharmacy University of Sevilla 41012 Sevilla Spain e-mail: [email protected] ISBN 978-3-642-05664-2 ISBN 978-3-662-07656-9 (eBook) DOI 10.1007/978-3-662-07656-9 Library of Congress Cataloging-in-Publication Data applied for A catalog record for this book is available from the Library of Congress. Bibliographic information published by Die Deutsche Bibliothek Die Deutsche Bibliothek lists this publicati an in the Deutsche Nationalbibliographie; detailed bibliographic data is available in the Internet at http://dnb.ddb.de This work is subject ta copyright. AII rights are reservod, whether the whole or part of the material is concerned, specifically the rights of translation, reprinting, reuse of iIIustrations, recitation, broadcasting, reproduction an microfilm ar in any other way, and storage in data banks. Duplication of this publication ar parts thereofis permit ted only under the provisions of the German Copyright Law of September 9, 1965, in ils current version, and per missions for use must always be obtained from Springcr-Vcrlag Berlin Heiddhcrg GmbH. Violations are liable for prosecution under the German Copyright Law. http://www.springer.de © Springer-Verlag Berlin Heidelberg 2004 Originally published by Springer-Vcrlag lkrlin Hcidclbcrg New York in 2004 The use of general descriptive names, registered names, trademarks, etc. in this publication does not imply, even in the absence of a specific statement, that such names are exempt from the relevant protective laws and regulations and therefore free for general use. -

Life at Low Water Activity
Published online 12 July 2004 Life at low water activity W. D. Grant Department of Infection, Immunity and Inflammation, University of Leicester, Maurice Shock Building, University Road, Leicester LE1 9HN, UK ([email protected]) Two major types of environment provide habitats for the most xerophilic organisms known: foods pre- served by some form of dehydration or enhanced sugar levels, and hypersaline sites where water availability is limited by a high concentration of salts (usually NaCl). These environments are essentially microbial habitats, with high-sugar foods being dominated by xerophilic (sometimes called osmophilic) filamentous fungi and yeasts, some of which are capable of growth at a water activity (aw) of 0.61, the lowest aw value for growth recorded to date. By contrast, high-salt environments are almost exclusively populated by prokaryotes, notably the haloarchaea, capable of growing in saturated NaCl (aw 0.75). Different strategies are employed for combating the osmotic stress imposed by high levels of solutes in the environment. Eukaryotes and most prokaryotes synthesize or accumulate organic so-called ‘compatible solutes’ (osmolytes) that have counterbalancing osmotic potential. A restricted range of bacteria and the haloar- chaea counterbalance osmotic stress imposed by NaCl by accumulating equivalent amounts of KCl. Haloarchaea become entrapped and survive for long periods inside halite (NaCl) crystals. They are also found in ancient subterranean halite (NaCl) deposits, leading to speculation about survival over geological time periods. Keywords: xerophiles; halophiles; haloarchaea; hypersaline lakes; osmoadaptation; microbial longevity 1. INTRODUCTION aw = P/P0 = n1/n1 ϩ n2, There are two major types of environment in which water where n is moles of solvent (water); n is moles of solute; availability can become limiting for an organism. -

Extracellular Hydrolases Producing Haloarchaea from Marine Salterns at Okhamadhi, Gujarat, India
Int.J.Curr.Microbiol.App.Sci (2016) 5(11): 51-64 International Journal of Current Microbiology and Applied Sciences ISSN: 2319-7706 Volume 5 Number 11 (2016) pp. 51-64 Journal homepage: http://www.ijcmas.com Original Research Article http://dx.doi.org/10.20546/ijcmas.2016.511.006 Extracellular Hydrolases producing Haloarchaea from Marine Salterns at Okhamadhi, Gujarat, India Bhavini N. Rathod1, Harshil H. Bhatt2 and Vivek N. Upasani1* 1Department of Microbiology, M.G. Science Institute, Ahmedabad- 380009, India 2Kadi Sarva Vishwavidyalay Gandhinagar-23, India *Corresponding author ABSTRACT Haloarchaea thrive in hypersaline environments such as marine salterns, saline soils, soda lakes, salted foods, etc. The lysis of marine phyto- and zoo-planktons such as algae, diatoms, shrimps, purple and green bacteria, fish, etc. releases K e yw or ds biopolymers namely cellulose, starch, chitin, proteins, lipids, etc. in the saline Extremozymes , ecosystems. The chemorganotrophic haloarchaea therefore, need to produce haloarchaea, hydrolytic enzymes to utilize these substrates. However, the raw solar salt used for hydrolases, preservation can cause spoilage of foods due to the growth of halobacteria leading Halobacterium, to economic loss. We report here the isolation and identification of extracellular Haloferax, hydrolases (substrates casein, gelatin, starch, and Tweens: 20, 60, 40, 80) Halopetinus, producing haloarchaea isolated from the salt and brine samples collected from Okhamadhi marine salterns at Okhamadhi, Gujarat, India. Morphological, -

The Role of Stress Proteins in Haloarchaea and Their Adaptive Response to Environmental Shifts
biomolecules Review The Role of Stress Proteins in Haloarchaea and Their Adaptive Response to Environmental Shifts Laura Matarredona ,Mónica Camacho, Basilio Zafrilla , María-José Bonete and Julia Esclapez * Agrochemistry and Biochemistry Department, Biochemistry and Molecular Biology Area, Faculty of Science, University of Alicante, Ap 99, 03080 Alicante, Spain; [email protected] (L.M.); [email protected] (M.C.); [email protected] (B.Z.); [email protected] (M.-J.B.) * Correspondence: [email protected]; Tel.: +34-965-903-880 Received: 31 July 2020; Accepted: 24 September 2020; Published: 29 September 2020 Abstract: Over the years, in order to survive in their natural environment, microbial communities have acquired adaptations to nonoptimal growth conditions. These shifts are usually related to stress conditions such as low/high solar radiation, extreme temperatures, oxidative stress, pH variations, changes in salinity, or a high concentration of heavy metals. In addition, climate change is resulting in these stress conditions becoming more significant due to the frequency and intensity of extreme weather events. The most relevant damaging effect of these stressors is protein denaturation. To cope with this effect, organisms have developed different mechanisms, wherein the stress genes play an important role in deciding which of them survive. Each organism has different responses that involve the activation of many genes and molecules as well as downregulation of other genes and pathways. Focused on salinity stress, the archaeal domain encompasses the most significant extremophiles living in high-salinity environments. To have the capacity to withstand this high salinity without losing protein structure and function, the microorganisms have distinct adaptations. -

Origin of the Cell Nucleus, Mitosis and Sex: Roles of Intracellular Coevolution Thomas Cavalier-Smith*
Cavalier-Smith Biology Direct 2010, 5:7 http://www.biology-direct.com/content/5/1/7 RESEARCH Open Access Origin of the cell nucleus, mitosis and sex: roles of intracellular coevolution Thomas Cavalier-Smith* Abstract Background: The transition from prokaryotes to eukaryotes was the most radical change in cell organisation since life began, with the largest ever burst of gene duplication and novelty. According to the coevolutionary theory of eukaryote origins, the fundamental innovations were the concerted origins of the endomembrane system and cytoskeleton, subsequently recruited to form the cell nucleus and coevolving mitotic apparatus, with numerous genetic eukaryotic novelties inevitable consequences of this compartmentation and novel DNA segregation mechanism. Physical and mutational mechanisms of origin of the nucleus are seldom considered beyond the long- standing assumption that it involved wrapping pre-existing endomembranes around chromatin. Discussions on the origin of sex typically overlook its association with protozoan entry into dormant walled cysts and the likely simultaneous coevolutionary, not sequential, origin of mitosis and meiosis. Results: I elucidate nuclear and mitotic coevolution, explaining the origins of dicer and small centromeric RNAs for positionally controlling centromeric heterochromatin, and how 27 major features of the cell nucleus evolved in four logical stages, making both mechanisms and selective advantages explicit: two initial stages (origin of 30 nm chromatin fibres, enabling DNA compaction; and firmer attachment of endomembranes to heterochromatin) protected DNA and nascent RNA from shearing by novel molecular motors mediating vesicle transport, division, and cytoplasmic motility. Then octagonal nuclear pore complexes (NPCs) arguably evolved from COPII coated vesicle proteins trapped in clumps by Ran GTPase-mediated cisternal fusion that generated the fenestrated nuclear envelope, preventing lethal complete cisternal fusion, and allowing passive protein and RNA exchange.