UNIVERSITY of CALIFORNIA Los Angeles PET Imaging of Nucleoside Metabolism for Individualized Therapy a Dissertation Submitted In
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Free Press Vol. 40, Issue No. 3, 09-22-2008
University of Southern Maine USM Digital Commons Free Press, The, 1971- Student Newspapers 9-22-2008 The Free Press Vol. 40, Issue No. 3, 09-22-2008 Matt Dodge University of Southern Maine Follow this and additional works at: https://digitalcommons.usm.maine.edu/free_press Recommended Citation Dodge, Matt, "The Free Press Vol. 40, Issue No. 3, 09-22-2008" (2008). Free Press, The, 1971-. 69. https://digitalcommons.usm.maine.edu/free_press/69 This Book is brought to you for free and open access by the Student Newspapers at USM Digital Commons. It has been accepted for inclusion in Free Press, The, 1971- by an authorized administrator of USM Digital Commons. For more information, please contact [email protected]. the free pressVolume 40, Issue no. 03 September 22, 2008 U S M Geing there, half the bale As Bedford Street continues to grow, new worries arise over parking David O’Donnell parking space before class on kinks at the start. The Free Press Tuesday. So when he returned published complaints at the time News Editor on Thursday, he made a point of that some drivers found the flow showing up a little earlier, only of traffic to be counter-intuitive, Alex Wilbur, to find an even bleaker situation. leaving some stuck in circles. a third-year He ended up 20-minutes late for More annoyingly, a computerized Linguistics stu- a sign language class. exit gate could sometimes seem dent, was surprised Around the same time, sopho- to channel the defiant H.A.L. when it took him more Business major Ashleigh from 2001: A Space Odyssey and about five Tatarcyk was finishing an un- reject perfectly good ID cards of min- successful lap around campus in people attempting to pass. -

Fall 2014 What’S New? New Minor: Iranian Studies the Near Eastern Languages and Cultures Department Has Established a New Minor in Iranian Studies
-15 ii UCLA Schedule of Classes Fall 2014 What’s New? New Minor: Iranian Studies The Near Eastern Languages and Cultures department has established a new minor in Iranian Studies. The minor gives stu- dents the opportunity to study Iranian languages and civilization while majoring in a different field of study. Introductory courses in the minor provide a foundation in Iranian history, languages, and religions, as well as Persian languages and literature. Other courses in the minor enable students to study particular areas of interest in greater depth. New Minor: Teaching Secondary Mathematics The Teaching Secondary Mathematics minor is designed for students majoring in fields other than mathematics who plan to teach secondary mathematics after graduation. The minor provides recognition for completion of prerequisite coursework for the Joint Mathematics Education Program and prepares students for topics covered on the California Subject Examination for Teachers (CSET) Mathematics test. Instructional Enhancement Initiative (IEI) Fee Billing Change Effective Fall 2014, the IEI fee is converted to a campus-based flat fee and charged to all undergraduate students. Previously the fee was unit-based and billed later in the quarter, based on enrollment in courses as of the fourth week of the quarter. Some students were not aware of the charge, and for some it impacted their ability to enroll in subsequent quarters or impeded their ability to graduate. With the new billing timing, students will be better informed of the fee and the fee can be included in finan- cial aid packages. This is not a fee increase. The fee generates the same amount of revenue as it did previously. -

Of Kappa Kappa- Gamma Fall 1968
OF KAPPA KAPPA- GAMMA FALL 1968 •• And God said~ Let there be light: and there was light.!!!! (Genesis 1:3) Light from that first dawn continues to bless God's universe. The light of sun and moon and stars, electric light illuminating the lives of millions, or the light of the single candle you hold-all dispel darkness. In time, the darkness of ignorance is dispelled by the light of knowledge. One such significant moment of enlightenment which occurred a hundred years ago opened wide the doors of institutions of higher learning formerly closed to women. Seeking to improve their new opportunities for education, some of those early college girls established the first women's Greek letter societies. The meetings they held were exercises in improvement. They prepared and read literary papers; they tried out musical and artistic talents; they debated and practiced oratory. The first reproof on record administered by members to a fellow member was reserved for the girl whose literary paper was judged not up to standards. Today when Kappas light candles in Kappa ceremonies, they do so to sym bolize the light they have promised to cherish throughout their lives. The lighted candles memorialize lives of women in all the Kappa chapters, since 1870, and the promises they make to uphold truth and goodness, faith and honor and reverence for God and His creations. "The power of our light is great, for better or worse. It is greater when we band together as in a sisterhood than when we stand alone." ( -Dr. Doris Seward, at the Candlelight Dinner, 1968 Biennial Convention.) OF KAPPA KAPPA GAMMA The first coUege women's magazine. -

The 3Rd Year Gazette April 2021 the 3Rd Year Gazette
The 3rd Year Gazette April 2021 The 3rd year Gazette I am procrastinating, you are procrastinating, he is procrastinating, she is procrastinating, we are procrastinating, they are procrastinating ….. By James Corbett Procrastination is the action of delaying or postponing something. The reason I chose to write about this today is because like most people I procrastinate but I guess it has helped with coming up with an idea for this gazette. Although on the article that "William Sydnor" wrote; he said that the student that procrastinated still got a fair grade I still believe that procrastination is the key to being a failure, eventually, it will catch up to you. In order to be successful one must manage time well. In my opinion, I think procrastination has a few stages which goes something like this: Stage 1) Having a plan all laid out to minimise workload. Stage 2) Then you usually find something else to do to get instant gratification. This is a stage that usually makes you feel good. Stage 3) But then all of a sudden you wake up one day and realise, oh no the project is due in two days. This is when you start to panic and just accept the fact that you have to do the work and realise it would've been better if you followed through on stage one. Why procrastination isn't about laziness. When we procrastinate, we’re not only aware that we’re avoiding the task in question, but also that doing so is probably a bad idea. And yet, we do it anyway. -

2017 Annual Report
2017 ANNUAL REPORT DIGNITY GROWTH TRANSFORMATION JOIN US BOARD OF DIRECTORS As a part of the SOIL community, you Melinda Miles are helping to create change every day. Chairperson Join us as we bring our vision to life. Jessica Covell Vice Chair David Reese Email: [email protected] Secretary Web: www.oursoil.org Robert McLaughlin Twitter: @SOILHaiti Treasurer Facebook: SOILHaiti Sasha Kramer Instagram: @SOILHaiti Executive Director Nick Preneta Visit: Deputy Director Northern Haiti: Rue 6 Nationale, Limonade Moira Duvernay Port-au-Prince: Rue A. Martial, Delmas 33 Cathy Ellenbogen Dignity | Transformation | Growth Lavarice Gaudin Ingrid Henrys Photo Credit: Claire Pask Jean Michel Petit Bois Dear friends, In 2006 when SOIL was founded in northern Haiti, we were determined to work in respect and solidarity with communities to determine what solutions we could co-create that would effect real, lasting change. Rather than coming in with a preformed idea and simply implementing it, we chose to engage in open-ended conversations about what these communities needed most. Over the past decade we have found that our quiet voices have started to echo around the world, growing into a passionate, vibrant conversation about how best to build a sanitation solution that works for everyone. Today, we are humbled and proud to see SOIL’s work held up as a success to be emulated globally. Our vision for a sustainable sanitation future is catching on and gaining momentum, and the SOIL team is still on the front lines – pushing for solutions that prioritize community empowerment, sustainable impact, and environmental restoration. We thank you – our friends, family, staff, supporters, and fellow dreamers – for being a continual source of inspiration, for pushing us to stay true to our commitments, and for making it possible for SOIL to grow into the international force for change that we are today. -
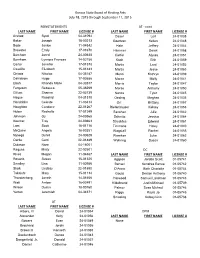
Last Name First Name License # Last Name First Name
Kansas State Board of Healing Arts July 18, 2015 through September 11, 2015 REINSTATEMENTS AT - cont. LAST NAME FIRST NAME LICENSE # LAST NAME FIRST NAME LICENSE # Arshad Syed 04-31094 Dauer Lori 24-01038 Baker Joseph 18-00213 Goertzen Kellen 24-01048 Bode Janice 11-04342 Hale Jeffrey 24-01044 Brewster Emily 17-01670 Hammer Derek 24-01058 Burcham Jared 24-00638 Kieffer Alyssa 24-01051 Burnham Lynnore Frances 14-02156 Koch Erin 24-01059 Carter Jennifer 17-01578 Martin Lani 24-01052 Caudillo Elizabeth 15-01443 Martin Jesse 24-01063 Chitaia Nikoloz 04-35187 Mena Kathryn 24-01039 Dahlstrom Hugo 17-02686 Moran Molly 24-01041 Eboh Rhanda Marie 04-33517 Morris Taylor 24-01047 Ferguson Rebecca 05-36269 Morse Anthony 24-01050 Gillum Shanna 22-03739 Nance Tyler 24-01045 Haque Raashid 04-31310 Oeding Meghan 24-01035 Hendricks Celeste 11-03414 Orr Brittany 24-01057 Houghton Candace 22-01267 Riefenhauser Kelsey 24-01053 Huber Rachelle 17-01249 Sanchez Julie 24-01042 Johnson Oji 24-00565 Schmitz Jessica 24-01064 Koerner Troy 24-00663 Streckfus Edward 24-01061 Lam Scott 16-01716 Timmons Haley 24-01040 McQuinn Angela 18-00351 Wagstaff Rachel 24-01055 Nyaega Durell 24-00629 Warnken Luke 24-01062 Oertle Carri 22-01889 Wehring Susan 24-01060 Quiason Nora 04-16001 Ragusa Misty 22-02501 DC Rives Megan 11-04467 LAST NAME FIRST NAME LICENSE # Rosario Susan 16-01325 Aggson Jordan Scott 01-05741 Smalley Lisa 11-02666 Bensel Kendrea Renee 01-05742 Stark Lindsay 22-01890 D'Anna Beth Charlotte 01-05744 Takaishi Mary 15-01134 Gould Dexton Anthony 01-05740 Thorstenberg Janeth 14-01656 Harwood Samuel Landman 01-05743 Watt Amber 16-02491 Hildebrand Justin Michael 01-05739 Wilson Dustin 18-00760 Palmer Sean Michael 01-05746 Yerton Jeremiah 04-34371 Riggs Kayla Jo 01-05745 Smading Kelsey Linn 01-05738 AT LAST NAME FIRST NAME LICENSE # Albers, III David 24-01054 DPM Alexander Kelly 24-01056 LAST NAME FIRST NAME LICENSE # Bowers Evan 24-01049 None Brouhard Jordin 24-01036 Brown III Kenneth 24-01065 Daellenbach Danae 24-01043 Dangerfield, Jr. -

Analysis of the Spanish Subtitling of the British Television Show 'Skins'
PONTIFICIA UNIVERSIDAD CATÓLICA DEL ECUADOR FACULTAD DE COMUNICACIÓN, LINGÜÍSTICA Y LITERATURA ESCUELA DE LINGÜÍSTICA DISERTACIÓN PREVIA A LA OBTENCIÓN DEL TÍTULO DE LICENCIADO EN LINGÜÍSTICA APLICADA A LA TRADUCCIÓN “Analysis of the Spanish subtitling of the British television show “Skins”: evaluation and alternative translation DIEGO ALBERTO GOMEZJURADO ÁVILA DIRECTORA: MTR. MILICA DRAGOSAVLJEVICH QUITO, 2014 TABLE OF CONTENTS ACKNOWLEDGEMENT .................................................................................................................... v ABSTRACT........................................................................................................................................ vi INTRODUCTION .............................................................................................................................. vii CHAPTER 1:...................................................................................................................................... 1 THEORETICAL AND CONCEPTUAL FRAMEWORK ...................................................................... 1 1.1. Background............................................................................................................................... 1 1.2. Linguistics ................................................................................................................................. 2 1.2.1. Phonetics and Phonology ................................................................................................. 2 1.2.2. Morphology -

Kansas Alumni Magazine
JAYHAWK GENERATIONS 2001 CALENDAR HOOPS GUIDE h h h NO. 6,2000 $5 Settle* into the Kansas basketball ttadiuo U mere -Ml_ f II i • Jc/ vy I I I mt SS 1-800-645-5409 Shop -'an-ysai four locations i Westridge Mall, Topek 9548 Antioch, Overland Park I Town Center Plaza, Leawood KANSAS ALUMNI CONTENTS e Magazine FEATURES 20 IRS I WORD Hail to the Chiefs FT THE CHORUS The American Presidency Series helped the dters from readers University Press of Kansas establish a name for ON THE BOULEVARD itself. Now, after 30 years, the esteemed series is Schedules of KU events running out of presidents to profile, but its influence JAYHAWK WALK endures. Smoking grass, Iron Chefs and more By Steven Hill LLTOPICS 'ews and notes, including Gilded Age art, 24 sea census and more How We Play the Game SPORTS nnball flirts with a mid- Roy Williams has found a home at KU, and the ason turnaround and University has found a coach who reminds us what asketball gears up it means to be afayhawk. OREAD READER By Chris Lazzarino Patrick Quinn looks at a singular pluralist: FDR Cover photograph by Wally Emerson OREAD WRITER * great teacher takes on a eat Dane 32 ASSOCIATION NEWS Woodward scholars, Halls of Academe omecoming and more Why is KU building new scholarship halls when ASS()( IATION many universities have given up on cooperative CALENDAR housing? Because students and alumni would The latest on chapter and have it no other way. Kill' events JAY1IAWK By Steven Hill GENERATIONS '"Icome to the Hill ASS NOTES c 'ram classmates 4ORY iths in the KU family SCHOOLWORK Page 32 /6 HAIL TO OLD KU Birth of a bird VOLUME 98 NO. -

Foundations & Corporate Donors
$1,000 to $2,499 $200 to $499 Foundations & AARP Maine Aetna Foundation Matching Gift Program Corporate Donors Anonymous Apple, Inc. Bangor Savings Bank Flaherty's Financial Services, LLC $15,000 and up The Boulos Company Ginn Foundation Anonymous Coffee By Design No Umbrella Onion Foundation CycleMania Rebel Cheesesteaks Robert G. and Jane V. Engel Foundation, Inc. Temple Beth El $7,500 to $10,000 Friends and Family of Great Diamond Island Terrence J. DeWan & Associates Evergreen Credit Union Greater Portland NEMBA IDEXX Laboratories, Inc. Harvard Pilgrim Health Care Foundation $100 to $199 L.L. Bean, Inc. Jade Integrated Health Allspeed Cyclery & Snow Machias Savings Bank Arkatechture $5,000 to $7,499 Norway Savings Bank Maine Initiatives Clark Insurance Retail Business Services MEMIC Martin's Point Health Care The Laura and Sam Rigby Family Fund Morgan Stanley Old Bug Light Charitable Foundation Thanksgiving Day Four Miler (TDFM) Owen Haskell Inc. WEX Inc. Rose Foods $500 to $999 Southern Maine Conservation Collaborative $2,500 to $4,999 Aristelle Teleflex Foundation 2DineIn LLC The Belayer Fund Title Loans Express Allagash Brewing Co. Buena Vista Landscaping Athletic Brewing Company Cornerstone Financial Planning Up to $99 AVANGRID Foundation CPort Credit Union AmazonSmile Foundation Bissell Brothers Brewing Co. Down East Group at Morgan Stanley JustGive David R. Kerr, D.D.S. / Maine Integrative DrummondWoodsum Kids Movement Project Dentistry Flatbread Company Maine State Employees Combined Developers Collaborative Float Harder Charitable -

Place Bib Name Time Age Gender 1 1718 Brian Walter 17:05 48 M 2
Place Bib Name Time Age Gender 1 1718 Brian Walter 17:05 48 M 2 1747 Kevin McGuire 17:33 49 M 3 1214 Taylor Jennings 18:08 25 M 4 1563 Michael Carter 18:27 19 M 5 1210 Andrew Barbera 18:51 17 M 6 1702 Trevor Stewart 19:03 42 M 7 1551 Vince Baranauskas 19:05 36 M 8 1558 Mark Bunker 19:46 34 M 9 1732 Michael Stowe 19:56 45 M 10 1239 Landon Bell 19:59 16 M 11 1559 Alistair Bushey 20:25 13 M 12 1231 Erik Olsen 20:27 47 M 13 1237 Nick Stillwell 20:29 14 M 14 1742 Sophia Link 20:39 14 F 15 1570 Ken Coddington 20:51 55 M 16 1235 Rob Semtner 20:54 42 M 17 1213 Mitchell Devenport 20:57 22 M 18 1671 Matthew McPherson 21:00 13 M 19 1230 Christian Shushok 21:00 14 M 20 1741 Emily Fitch 21:19 14 F 21 1658 Matt Lyerly 21:20 M 22 1726 Marcia Wright 21:37 37 F 23 1242 Tim Crockett 21:38 43 M 24 1604 Sean Heuser 21:43 32 M 25 1234 Brady Semtner 21:47 12 M 26 1662 Cortney Martin 21:52 48 F 27 1233 Casey Phlegar 21:56 22 F 28 1723 Kaitlynn Wolfe 22:01 13 F 29 1241 Lane Holbert 22:10 51 M 30 1219 Randy Burrier 22:14 56 M 31 1232 Kamryn Phlegar 22:18 16 F 32 1648 Sarah Langlinais 22:20 40 F 33 1647 Brandon Langlinais 22:23 42 M 34 1211 George Barbera 22:26 48 M 35 1636 Tom Inzana 22:30 62 M 36 1238 Barry Robert 22:34 55 M 37 1709 Matthew Tolbert 22:54 39 M 38 1687 Trish Richardson 23:04 54 F 39 1224 Dennis Grove 23:21 41 M 40 1659 Marc MaGruder 23:30 40 M 41 1746 John Wilson 23:30 57 M 42 1670 David McPherson 23:36 44 M 43 1602 Travis Hawley 23:38 33 M 44 1700 Ben Sternfeld 23:43 21 M 45 1706 Sydney Sumner 23:43 21 F 46 1679 Daniel Overstreet 23:43 25 M 47 -

Open Zachs Thesis-Pdf.Pdf
THE PENNSYLVANIA STATE UNIVERSITY SCHREYER HONORS COLLEGE DEPARTMENT OF ENGLISH THE OBJECTIFICATION OF WOMEN IN FILM: A PSYCHOANALYTICAL READING ZACHARY WEIT SPRING 2014 A thesis submitted in partial fulfillment of the requirements for baccalaureate degrees in English and Spanish with honors in English Reviewed and approved* by the following: Iyunolu Osagie Associate Professor of Race and Ethnicity Studies Thesis Supervisor Marcy North Associate Professor of English Penn State Representative to the Folger Shakespeare Library Honors Adviser * Signatures are on file in the Schreyer Honors College. i ABSTRACT My thesis discusses the objectification of women in the films “Transformers,” “Transformers: Dark of the Moon,” and “Crash.” The two “Transformers” movies demonstrate the roles of gender and how the male gaze operates within a Hollywood action film. The director, Michael Bay, uses his female protagonists as a spectacle for the audience to consume; these female protagonists are in fact extraneous to the plot. “Crash,” directed by Paul Haggis, shows the racial side of objectification. A white cop sexually molests one of the leading black female characters. The police officer uses her as commodified object to antagonize her husband who can do nothing but helplessly watch. My thesis uses the works of Sigmund Freud, Ferdinand de Saussure, and Jacques Lacan to explore the process film directors use to objectify women. Sigmund Freud’s reading of Sophocles’ Oedipus Rex introduces his psychoanalytic concept of the Oedipus complex, where a young boy has sexual feelings for his mother and jealous hatred towards his father. Through his research of dreams, Freud then uses E.T.A. -

Seniors Win COMMENCEMENT
MAY 2011 VOLUME 59 NUMBER 6 by Howard Heaton, President, Class of 2011 Sunshine. Hail. Sunshine. Wind. Snow. Sunshine. Sunshine and Snow. We have become accustomed to the strange weather of Spangle. And through it all this year has brought many laughs and great times. There was the handshake, fall picnic, class parties, mission trips, and more. Cherish these memories because they and the friends you make here can last a lifetime. In these last few days as graduation draws near, the days will come more and more quickly. The seniors drag their feet going to class and count down the days until graduation. The juniors are preparing to take on their new role. The sophomores ready themselves to be upperclassmen while the freshmen soak it all in for the first time. It’s been an honor to learn and be a part of UCA. Now the task remains to decide which memories we want to take with us. UCA is more than just a bunch of buildings and classrooms. It’s the people, the lessons you learn, and the great memories you make along the way. We seniors have had a blast. But don’t kid yourself. There will be long nights of homework and running for Soulé, but it is all worth it in the end. Juniors, be ready. Next year is going to flash before your eyes and before you know it you’ll be here in our shoes. Let’s cherish these remaining days and continue to let God be number one on campus. Seniors win Bloomsday School Challenge 2011 AIM: May everything we do or say be for the glory of God.