Mapping the Conformational Dynamics of E-Selectin Upon Interaction with Its Ligands
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Regulation of Apoptosis by P53-Inducible Transmembrane Protein Containing Sushi Domain
1193-1200.qxd 24/9/2010 08:07 Ì ™ÂÏ›‰·1193 ONCOLOGY REPORTS 24: 1193-1200, 2010 Regulation of apoptosis by p53-inducible transmembrane protein containing sushi domain HONGYAN CUI, HIROKI KAMINO, YASUYUKI NAKAMURA, NORIAKI KITAMURA, TAKAFUMI MIYAMOTO, DAISUKE SHINOGI, OLGA GODA, HIROFUMI ARAKARA and MANABU FUTAMURA Cancer Medicine and Biophysics Division, National Cancer Center Research Institute, 5-1-1 Tsukiji, Chuo-ku, Tokyo 104-0045, Japan Received January 14, 2010; Accepted March 30, 2010 DOI: 10.3892/or_00000972 Abstract. The tumor suppressor p53 is a transcription factor repair, apoptosis, and anti-angiogenesis (1,2). The p21/waf1 that induces the transcription of various target genes in gene is considered to be one of the most important p53 target response to DNA damage and it protects the cells from genes because it is essential for p53-dependent cell cycle malignant transformation. In this study, we performed cDNA arrest at G1. p53R2, which supplies nucleotides for DNA microarray analysis and found that the transmembrane protein synthesis, facilitates the repair of DNA damage. Several containing sushi domain (TMPS) gene, which encodes a mitochondrial proteins including BAX, Noxa, and p53- putative type I transmembrane protein, is a novel p53-target regulated apoptosis-inducing protein 1 (p53AIP1) contribute gene. TMPS contains a sushi domain in the extracellular to the release of cytochrome c from mitochondria. Other region, which is associated with protein-protein interaction. proteins such as Fas/ApoI and unc-5 homolog B (UNC5B) TMPS expression is induced by endogenous p53 under geno- are also associated with apoptosis. Brain-specific angio- toxic stress in several cancer cell lines. -

Spatially Conserved Motifs in Complement Control Protein
ARTICLE https://doi.org/10.1038/s42003-019-0529-9 OPEN Spatially conserved motifs in complement control protein domains determine functionality in regulators of complement activation-family proteins 1234567890():,; Hina Ojha1, Payel Ghosh2, Hemendra Singh Panwar 1, Rajashri Shende1, Aishwarya Gondane2, Shekhar C. Mande 3,4 & Arvind Sahu 1 Regulation of complement activation in the host cells is mediated primarily by the regulators of complement activation (RCA) family proteins that are formed by tandemly repeating complement control protein (CCP) domains. Functional annotation of these proteins, how- ever, is challenging as contiguous CCP domains are found in proteins with varied functions. Here, by employing an in silico approach, we identify five motifs which are conserved spatially in a specific order in the regulatory CCP domains of known RCA proteins. We report that the presence of these motifs in a specific pattern is sufficient to annotate regulatory domains in RCA proteins. We show that incorporation of the lost motif in the fourth long-homologous repeat (LHR-D) in complement receptor 1 regains its regulatory activity. Additionally, the motif pattern also helped annotate human polydom as a complement regulator. Thus, we propose that the motifs identified here are the determinants of functionality in RCA proteins. 1 Complement Biology Laboratory, National Centre for Cell Science, S. P. Pune University campus, Pune 411007, India. 2 Bioinformatics Centre, S. P. Pune University, Pune 411007, India. 3 Structural Biology Laboratory, National Centre for Cell Science, S. P. Pune University campus, Pune 411007, India. 4Present address: Council of Scientific and Industrial Research (CSIR), Anusandhan Bhawan, 2 Rafi Marg, New Delhi 110001, India. -

And Mouse CR1 and CR2 Comparative Functional Evolution of Human
Comparative Functional Evolution of Human and Mouse CR1 and CR2 Amanda C. Jacobson and John H. Weis This information is current as J Immunol 2008; 181:2953-2959; ; of September 26, 2021. doi: 10.4049/jimmunol.181.5.2953 http://www.jimmunol.org/content/181/5/2953 Downloaded from References This article cites 85 articles, 54 of which you can access for free at: http://www.jimmunol.org/content/181/5/2953.full#ref-list-1 Why The JI? Submit online. http://www.jimmunol.org/ • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication *average by guest on September 26, 2021 Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2008 by The American Association of Immunologists All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. Comparative Functional Evolution of Human and Mouse CR1 and CR21,2 Amanda C. Jacobson and John H. Weis3 he complement cascade is regulated by a series of pro- The C3 complement convertases are the targets of many of teins that inhibit complement convertase activity. the complement regulatory proteins. For example, decay accel- T These regulatory proteins, most of which possess bind- eration factor (DAF) enhances the decay of complement con- ing sites for C3b and/or C4b, can be roughly divided into two vertases by binding to C3b (5, 6). -

Loss of Ancestral N-Glycosylation Sites in Conserved Proteins During Human Evolution
INTERNATIONAL JOURNAL OF MOLECULAR MEDICINE 36: 1685-1692, 2015 Loss of ancestral N-glycosylation sites in conserved proteins during human evolution DONG SEON KIM, DONGJIN CHOI and YOONSOO HAHN Department of Life Science, Research Center for Biomolecules and Biosystems, Chung-Ang University, Seoul 156-756, Republic of Korea Received June 25, 2015; Accepted October 1, 2015 DOI: 10.3892/ijmm.2015.2362 Abstract. N-linked protein glycosylation is involved in We hypothesized that the losses of certain ancestrally various biological processes, such as protein quality control conserved N-glycosylation sites during evolution may and adhesion or signaling among cells. The loss of ancestrally have been involved in the acquisition of novel human conserved N-glycosylation sites may result in the evolu- phenotypes. The loss of N-glycosylation often disrupts the tion of protein structure and function. In the present study, normal function of proteins due to improper folding, traf- a mouse glycoproteome dataset and mammalian proteome ficking, or activity of the proteins (6,7). A proteome-wide data were assessed to identify 40 ancestral N-glycosylation analysis of non-synonymous single-nucleotide variations in sites in 37 proteins that disappeared during human evolu- the N-glycosylation motifs of human proteins revealed that tion since the last common ancestor of the Euarchonta 259 sites were lost because of missense substitutions, some (primates and treeshrews). The results showed that each of of which are involved in various diseases (8). Although loss the human proteins, CELSR1, ST3GAL5 and VSIG10, lost of a glycosylation modification usually results in disadvanta- an ancestrally conserved N-glycosylation site following geous phenotypes, some losses may be beneficial and fixed human-chimpanzee divergence. -

Supplementary Information
SUPPLEMENTARY INFORMATION 1. SUPPLEMENTARY FIGURE LEGENDS Supplementary Figure 1. Long-term exposure to sorafenib increases the expression of progenitor cell-like features. A) mRNA expression levels of PROM-1 (CD133), THY-1 (CD90), EpCAM, KRT19, and VIM assessed by quantitative real-time PCR. Data represent the mean expression value for a gene in each phenotypic type of cells, displayed as fold-changes normalized to 1 (expression value of its corresponding parental non-treated cell line). Expression level is relative to the GAPDH gene. Bars indicate standard deviation. Significant statistical differences are set at p<0.05. B) Immunocitochemical staining of CD90 and vimentin in Hep3B sorafenib resistant cell line and its parental cell line. C) Western blot analysis comparing protein levels in resistant Hu6 and Hep3B cells vs their corresponding parental cells lines. Supplementary Figure 2. Efficacy of gene silencing of IGF1R and FGFR1 and evaluation of MAPK14 signaling activation. IGF1R and FGFR1 knockdown expression 48h after transient transfection with siRNAs (50 nM), in non-treated parental cells and sorafenib-acquired resistant tumor derived cells was assessed by quantitative RT-PCR (A) and western blot (B). C) Activation status of MAPK14 signaling was evaluated by western blot analysis in vivo, in tumors with acquired resistance to sorafenib in comparison to non-treated tumors (right panel), as well as in in vitro, in sorafenib resistant cell lines vs parental non-treated. Supplementary Figure 3. Gene expression levels of several pro-angiogenic factors. mRNA expression levels of FGF1, FGF2, VEGFA, IL8, ANGPT2, KDR, FGFR3, FGFR4 assessed by quantitative real-time PCR in tumors harvested from mice. -

Molecular Evolution of the Human SRPX2 Gene That Causes Brain Disorders of the Rolandic and Sylvian Speech Areas
Edinburgh Research Explorer Molecular evolution of the human SRPX2 gene that causes brain disorders of the Rolandic and Sylvian speech areas Citation for published version: Royer, B, Soares, DC, Barlow, PN, Bontrop, RE, Roll, P, Robaglia-Schlupp, A, Blancher, A, Levasseur, A, Cau, P, Pontarotti, P & Szepetowski, P 2007, 'Molecular evolution of the human SRPX2 gene that causes brain disorders of the Rolandic and Sylvian speech areas', BMC Genetics, vol. 8, no. 72, pp. 1-9. https://doi.org/10.1186/1471-2156-8-72 Digital Object Identifier (DOI): 10.1186/1471-2156-8-72 Link: Link to publication record in Edinburgh Research Explorer Document Version: Publisher's PDF, also known as Version of record Published In: BMC Genetics General rights Copyright for the publications made accessible via the Edinburgh Research Explorer is retained by the author(s) and / or other copyright owners and it is a condition of accessing these publications that users recognise and abide by the legal requirements associated with these rights. Take down policy The University of Edinburgh has made every reasonable effort to ensure that Edinburgh Research Explorer content complies with UK legislation. If you believe that the public display of this file breaches copyright please contact [email protected] providing details, and we will remove access to the work immediately and investigate your claim. Download date: 24. Sep. 2021 BMC Genetics BioMed Central Research article Open Access Molecular evolution of the human SRPX2 gene that causes brain disorders of the Rolandic -

The Schizophrenia-Associated Gene, CSMD1, Encodes a Brain- Specific Complement Inhibitor
The Schizophrenia-Associated Gene, CSMD1, Encodes a Brain- Specific Complement Inhibitor The Harvard community has made this article openly available. Please share how this access benefits you. Your story matters Citation Baum, Matthew L. 2018. The Schizophrenia-Associated Gene, CSMD1, Encodes a Brain-Specific Complement Inhibitor. Doctoral dissertation, Harvard University, Graduate School of Arts & Sciences. Citable link http://nrs.harvard.edu/urn-3:HUL.InstRepos:41121210 Terms of Use This article was downloaded from Harvard University’s DASH repository, and is made available under the terms and conditions applicable to Other Posted Material, as set forth at http:// nrs.harvard.edu/urn-3:HUL.InstRepos:dash.current.terms-of- use#LAA The Schizophrenia-Associated Gene, CSMD1, Encodes a Brain-Specific Complement Inhibitor A dissertation presented by Matthew L Baum to The Division of Medical Sciences in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the subject of Neurobiology Harvard University Cambridge, Massachusetts June 2018 © 2018 Matthew L Baum All rights reserved. Dissertation Advisors: Drs. Beth Stevens and Steven A. McCarroll Matthew L Baum The Schizophrenia-Associated Gene, CSMD1, Encodes a Brain-Specific Complement Inhibitor Abstract Four threads of biological observation have recently converged into a theory in which over- exuberant synaptic pruning contributes to the pathogenesis of Schizophrenia: 1) post mortem, there is a reduced number of dendritic spines in the frontal cortex of individuals -
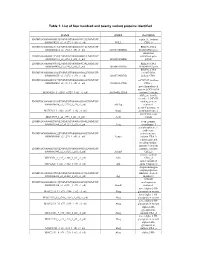
List of Four Hundred and Twenty Variant Proteins Identified
Table 1: List of four hundred and twenty variant proteins identified protein symbol description ENSMUSG00000058013|ENSMUST00000074733|ENSMUSP septin 11, isoform 00000074293_s2_e1277_1_rf0_c1_n0| NULL CRA_b ENSMUSG00000025176|ENSMUST00000081714|ENSMUSP RIKEN cDNA 00000080414_s2_e965_1_rf0_c1_n0| 0610010D20Rik 0610010D20 gene interferon ENSMUSG00000021208|ENSMUST00000044687|ENSMUSP stimulated gene 00000041712_s2_e851_1_rf0_c1_n0| 1810023F06Rik 12(b2) ENSMUSG00000036938|ENSMUST00000064324|ENSMUSP RIKEN cDNA 00000064498_s2_e740_1_rf0_c1_n0| 1810049H19Rik 1810049H19 gene RIKEN cDNA ENSMUSG00000061306|ENSMUST00000045402|ENSMUSP 1810073N04, 00000048675_s2_e3272_1_rf0_c1_n0| 1810073N04Rik isoform CRA_ ENSMUSG00000039879|ENSMUST00000020002|ENSMUSP mCG7517, isoform 00000020002_s2_e314_1_rf0_c1_n0| 3110003A17Rik CRA_c novel hypothetical protein LOC106894 M18C4209_1_s2543_e2729_1_rf1_c1_n0| A630042L21Rik isoform 2 variant ABI gene family, member 3 (NESH) ENSMUSG00000035258|ENSMUST00000048280|ENSMUSP binding protein 00000040830_s2_e2729_1_rf0_c1_n0| Abi3bp isoform 1 acetyl-Coenzyme A M17C923_1_s128_e1337_1_rf2_c1_n0| Acat2 acetyltransferase 2 novel beta actin MXC1985_1_s2_e593_1_rf1_c1_n0| Actb variant ENSMUSG00000029580|ENSMUST00000031564|ENSMUSP actin, gamma, 00000031564_s2_e1127_1_rf0_c1_n0| Actg cytoplasmic 1 acylphosphatase 1, erythrocyte ENSMUSG00000008822|ENSMUST00000008966|ENSMUSP (common) type, 00000008966_s2_e299_1_rf0_c1_n0| Acyp1 isoform CRA_b a disintegrin and metallopeptidase domain 9 (meltrin ENSMUSG00000031555|ENSMUST00000084032|ENSMUSP -

4 353 Skin Oral 1 B
A B Supplementary Figure S1: Differentially expressed piRNAs during skin and oral mucosal wound healing. (A) piRNA 0hr-1 0hr-3 0hr-2 24hr-2 24hr-1 24hr-3 5day-3 5day-1 5day-2 0hr-1 0hr-2 0hr-3 24hr-2 24hr-1 24hr-3 5day-1 5day-2 5day-3 profiles were obtained on mouse skin and oral mucosal (palate) wound healing time course (0hr, 24 hr, and 5 day). A total of 357 differentially expressed piRNA were identified during skin wound healing (Bonferroni adjusted P value <0.05). See Supplementary Table 1A for the full list. C (B) Five differentially expressed piRNA skin were identified during oral mucosal wound healing (P value <0.01, list presented in Supplementary Table 1B). Note: more 353 stringent statistical cut-off ((Bonferroni adjusted P value) yield 0 differentially expressed piRNA gene. (C) Venn diagram illustrates overlaps between differentially 4 expressedpiRNAsinskinandoral oral 1 mucosal wound healing. min max Supplementary Table S1a: Differentially expressed piRNAs in skin wound healing Mean StDev piRNA 0 hr 24 hr 5 day 0 hr 24 hr 5 day pVal adj P piR‐mmu‐15927330 5.418351 11.39746 10.799 0.34576 0.253492 0.154802 6.32E‐14 6.97E‐11 piR‐mmu‐49559417 5.301647 10.10777 9.816878 0.441719 0.222335 0.032479 3.95E‐12 4.36E‐09 piR‐mmu‐30053093 6.32531 11.26384 4.020902 0.280841 1.057847 0.178798 5.21E‐12 5.76E‐09 piR‐mmu‐29303577 5.15005 10.47662 9.52175 0.554877 0.26622 0.163283 1.53E‐11 1.69E‐08 piR‐mmu‐49254706 5.187673 10.19644 9.622374 0.520671 0.330378 0.191116 1.96E‐11 2.16E‐08 piR‐mmu‐49005170 5.415133 9.639725 9.565967 0.411507 0.281143 -

The Sez6 Family Inhibits Complement at the Level of the C3 Convertase
bioRxiv preprint doi: https://doi.org/10.1101/2020.09.11.292623; this version posted September 11, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. The Sez6 family inhibits complement at the level of the C3 convertase Wen Q. Qiu1#,, Shaopeiwen Luo1#, Stefanie A. Ma1, Priyanka Saminathan1, Herman Li1, Jenny Gunnersen2, Harris A. Gelbard1, Jennetta W. Hammond1* Author Affiliations: 1. Center for Neurotherapeutics Discovery, Department of Neurology, University of Rochester Medical Center, 601 Elmwood Avenue, Rochester NY 14642. 2. Department of Anatomy and Neuroscience and The Florey Institute of Neuroscience and Mental Health, University of Melbourne, Melbourne, VIC 3010, Australia. #Authors contributed equally to the work *Correspondence: Jennetta W. Hammond University of Rochester Dept. of Neurology, Center for Neurotherapeutics Discovery 601 Elmwood Avenue, Box 645 Rochester, NY 14642, USA [email protected] Keywords: Sez6L2, Sez6, Sez6L complement, classical pathway, alternative pathway, C3 convertase, Factor I cofactor, decay accelerating activity, C3b, C4b. bioRxiv preprint doi: https://doi.org/10.1101/2020.09.11.292623; this version posted September 11, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Abstract The Sez6 family consists of Sez6, Sez6L, and Sez6L2. Its members are expressed throughout the brain and have been shown to influence synapse numbers and dendritic morphology. -

Deimination Protein Profiles in Alligator Mississippiensis Reveal
ORIGINAL RESEARCH published: 28 April 2020 doi: 10.3389/fimmu.2020.00651 Deimination Protein Profiles in Alligator mississippiensis Reveal Plasma and Extracellular Vesicle-Specific Signatures Relating to Immunity, Metabolic Function, and Gene Regulation Michael F. Criscitiello 1,2, Igor Kraev 3, Lene H. Petersen 4 and Sigrun Lange 5* 1 Comparative Immunogenetics Laboratory, Department of Veterinary Pathobiology, College of Veterinary Medicine and Biomedical Sciences, Texas A&M University, College Station, TX, United States, 2 Department of Microbial Pathogenesis and Immunology, College of Medicine, Texas A&M Health Science Center, Texas A&M University, College Station, TX, United States, 3 Electron Microscopy Suite, Faculty of Science, Technology, Engineering and Mathematics, Open University, Milton Keynes, United Kingdom, 4 Department of Marine Biology, Texas A&M University at Galvestone, Galveston, TX, United States, 5 Tissue Architecture and Regeneration Research Group, School of Life Sciences, University of Westminster, London, United Kingdom Alligators are crocodilians and among few species that endured the Edited by: Cretaceous–Paleogene extinction event. With long life spans, low metabolic rates, Fabrizio Ceciliani, unusual immunological characteristics, including strong antibacterial and antiviral ability, University of Milan, Italy and cancer resistance, crocodilians may hold information for molecular pathways Reviewed by: underlying such physiological traits. Peptidylarginine deiminases (PADs) are a group of Marie-Claire Mechin, Université Toulouse III Paul calcium-activated enzymes that cause posttranslational protein deimination/citrullination Sabatier, France in a range of target proteins contributing to protein moonlighting functions in health Hai-peng Liu, Xiamen University, China and disease. PADs are phylogenetically conserved and are also a key regulator of *Correspondence: extracellular vesicle (EV) release, a critical part of cellular communication. -
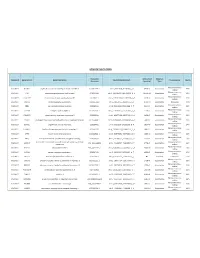
Somatic Mutations
SOMATIC MUTATIONS Transcript Amino Acid Mutation Sample ID Gene Symbol Gene Description Nucleotide (genomic) Consequence Mut % Accession (protein) Type Nonsynonymous PGDX11T ACSBG2 acyl-CoA synthetase bubblegum family member 2 CCDS12159.1 chr19_6141628_6141628_C_A 654A>D Substitution 46% coding Nonsynonymous PGDX11T ATR ataxia telangiectasia and Rad3 related CCDS3124.1 chr3_143721232_143721232_G_A 1451R>W Substitution 32% coding Nonsynonymous PGDX11T C1orf183 chromosome 1 open reading frame 183 CCDS841.1 chr1_112071336_112071336_C_T 224R>Q Substitution 22% coding PGDX11T CMYA5 cardiomyopathy associated 5 NM_153610 chr5_79063455_79063455_C_A 1037C>X Substitution Nonsense 34% Nonsynonymous PGDX11T CNR1 cannabinoid receptor 1 (brain) CCDS5015.1 chr6_88911646_88911646_C_T 23V>M Substitution 35% coding Nonsynonymous PGDX11T COL4A4 collagen; type IV; alpha 4 CCDS42828.1 chr2_227681807_227681807_G_A 227R>C Substitution 20% coding Nonsynonymous PGDX11T CYBASC3 cytochrome b; ascorbate dependent 3 CCDS8004.1 chr11_60877136_60877136_G_A 149R>C Substitution 34% coding Nonsynonymous PGDX11T DYRK3 dual-specificity tyrosine-(Y)-phosphorylation regulated kinase 3 CCDS30999.1 chr1_204888091_204888091_G_A 309V>I Substitution 44% coding Nonsynonymous PGDX11T ELMO1 engulfment and cell motility 1 CCDS5449.1 chr7_37239295_37239295_G_A 160T>M Substitution 24% coding Nonsynonymous PGDX11T FAM83H family with sequence similarity 83; member H CCDS6410.2 chr8_144884473_144884473_C_A 90G>C Substitution 53% coding Nonsynonymous PGDX11T KPTN kaptin (actin binding protein)