IBRAHIM ALDEAILEJ Phd.2013.Pdf
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Differentiation-Defective Phenotypes Revealed by Large-Scale Analyses of Human Pluripotent Stem Cells
Differentiation-defective phenotypes revealed by large-scale analyses of human pluripotent stem cells Michiyo Koyanagi-Aoia,1, Mari Ohnukia,1, Kazutoshi Takahashia, Keisuke Okitaa, Hisashi Nomab, Yuka Sawamuraa, Ito Teramotoa, Megumi Naritaa, Yoshiko Satoa, Tomoko Ichisakaa, Naoki Amanoa, Akira Watanabea, Asuka Morizanea, Yasuhiro Yamadaa,c, Tosiya Satod, Jun Takahashia,e, and Shinya Yamanakaa,f,2 aCenter for iPS Cell Research and Application, eDepartment of Biological Repair, Institute for Frontier Medical Sciences, and cInstitute for Integrated Cell- Material Sciences, Kyoto University, Kyoto 606-8507, Japan; bDepartment of Data Science, The Institute of Statistical Mathematics, Tokyo 190-8562, Japan; dDepartment of Biostatistics, Kyoto University School of Public Health, Kyoto 606-8501, Japan; and fGladstone Institute of Cardiovascular Disease, San Francisco, CA 94158 Contributed by Shinya Yamanaka, October 30, 2013 (sent for review September 13, 2013) We examined the gene expression and DNA methylation of 49 at least three passages. In addition, we analyzed the original somatic human induced pluripotent stem cells (hiPSCs) and 10 human cells, two human embryonic carcinoma cell (hECC) lines (NTera2 embryonic stem cells and found overlapped variations in gene cloneD1 and 2102Ep 4D3), and three cancer cell lines (HepG2, expression and DNA methylation in the two types of human MCF7, and Jurkat). pluripotent stem cell lines. Comparisons of the in vitro neural The mRNA microarray analyses (Fig. 1A) identified 61 probes fi differentiation of 40 hiPSCs and 10 human embryonic stem cells with signi cant differences in expression between hESCs and < showed that seven hiPSC clones retained a significant number of hiPSCs [t test, false discovery rate (FDR) 0.05]. -

Supporting Information
Supporting Information Fabbri et al. 10.1073/pnas.1702564114 a. b. GC 4X GCs 20x GC 4X GCs 4X Tonsil epithelium ICN1 CD20 DAPI Fig. S1. ICN1 is expressed in the B-cell fraction populating the mantle zone (M) of the germinal centers (GCs). (A) Double IF staining of ICN1 and AID in a rep- resentative GC in a human tonsil section. (B) Double IF staining of ICN1 and the B-cell–specific surface antigen CD20 at lower magnification (4×) in a human tonsil section. Fabbri et al. www.pnas.org/cgi/content/short/1702564114 1of10 Fig. S2. ICN1 expression analysis in a panel of primary CLL cases and PBMC. (A) IB analysis of ICN1 and control β-actin in a panel of 124 CLL PB primary CLL cases, (B) in primary NOTCH1–wild-type CLL cells treated with the γ-secretase inhibitor Compound E (CpE, 500 nM, 8 h) or control DMSO, and (C) in PBMC protein extracts and representative primary CLL cases expressing ICN1. Samples are color-coded based on the NOTCH1 mutational status [red, clonal NOTCH1 PEST-truncating events; orange, subclonal NOTCH1 PEST-truncating events; blue, RAG-mediated NOTCH1 translocation (83); and black, NOTCH1–wild-type]. Samples in gray were excluded from the analysis because of low quality of the protein lysate, low viability, or low leukemic representation. Color-coded arrows indicate cases subjected to RNA-Seq analysis: dark red denotes NOTCH1-mutated cases expressing ICN1; blue, NOTCH1–wild-type cases expressing ICN1; and green, ICN1− NOTCH1–wild-type cases. Abbreviations: MO+DL1, MO1043 cells cocultured on OP9-DL1 cells (54); s.e., short exposure; l.e., long exposure. -

Essential Genes and Their Role in Autism Spectrum Disorder
University of Pennsylvania ScholarlyCommons Publicly Accessible Penn Dissertations 2017 Essential Genes And Their Role In Autism Spectrum Disorder Xiao Ji University of Pennsylvania, [email protected] Follow this and additional works at: https://repository.upenn.edu/edissertations Part of the Bioinformatics Commons, and the Genetics Commons Recommended Citation Ji, Xiao, "Essential Genes And Their Role In Autism Spectrum Disorder" (2017). Publicly Accessible Penn Dissertations. 2369. https://repository.upenn.edu/edissertations/2369 This paper is posted at ScholarlyCommons. https://repository.upenn.edu/edissertations/2369 For more information, please contact [email protected]. Essential Genes And Their Role In Autism Spectrum Disorder Abstract Essential genes (EGs) play central roles in fundamental cellular processes and are required for the survival of an organism. EGs are enriched for human disease genes and are under strong purifying selection. This intolerance to deleterious mutations, commonly observed haploinsufficiency and the importance of EGs in pre- and postnatal development suggests a possible cumulative effect of deleterious variants in EGs on complex neurodevelopmental disorders. Autism spectrum disorder (ASD) is a heterogeneous, highly heritable neurodevelopmental syndrome characterized by impaired social interaction, communication and repetitive behavior. More and more genetic evidence points to a polygenic model of ASD and it is estimated that hundreds of genes contribute to ASD. The central question addressed in this dissertation is whether genes with a strong effect on survival and fitness (i.e. EGs) play a specific oler in ASD risk. I compiled a comprehensive catalog of 3,915 mammalian EGs by combining human orthologs of lethal genes in knockout mice and genes responsible for cell-based essentiality. -

Transcriptional Profiling Identifies the Lncrna PVT1 As a Novel Regulator of the Asthmatic Phenotype in Human Airway Smooth Muscle
Accepted Manuscript Transcriptional profiling identifies the lncRNA PVT1 as a novel regulator of the asthmatic phenotype in human airway smooth muscle Philip J. Austin, MSc, Eleni Tsitsiou, PhD, Charlotte Boardman, MD, Simon W. Jones, PhD, Mark A. Lindsay, PhD, Ian M. Adcock, PhD, Kian Fan Chung, MD PhD, Mark M. Perry, PhD PII: S0091-6749(16)30571-1 DOI: 10.1016/j.jaci.2016.06.014 Reference: YMAI 12203 To appear in: Journal of Allergy and Clinical Immunology Received Date: 5 April 2016 Revised Date: 24 May 2016 Accepted Date: 13 June 2016 Please cite this article as: Austin PJ, Tsitsiou E, Boardman C, Jones SW, Lindsay MA, Adcock IM, Chung KF, Perry MM, Transcriptional profiling identifies the lncRNA PVT1 as a novel regulator of the asthmatic phenotype in human airway smooth muscle, Journal of Allergy and Clinical Immunology (2016), doi: 10.1016/j.jaci.2016.06.014. This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain. ACCEPTED MANUSCRIPT 1 Transcriptional profiling identifies the lncRNA PVT1 as a novel 2 regulator of the asthmatic phenotype in human airway smooth muscle 3 4 Philip J. Austin MSc 1, Eleni Tsitsiou PhD 2, Charlotte Boardman MD 1, Simon W. -

Arnau Soler2019.Pdf
This thesis has been submitted in fulfilment of the requirements for a postgraduate degree (e.g. PhD, MPhil, DClinPsychol) at the University of Edinburgh. Please note the following terms and conditions of use: This work is protected by copyright and other intellectual property rights, which are retained by the thesis author, unless otherwise stated. A copy can be downloaded for personal non-commercial research or study, without prior permission or charge. This thesis cannot be reproduced or quoted extensively from without first obtaining permission in writing from the author. The content must not be changed in any way or sold commercially in any format or medium without the formal permission of the author. When referring to this work, full bibliographic details including the author, title, awarding institution and date of the thesis must be given. Genetic responses to environmental stress underlying major depressive disorder Aleix Arnau Soler Doctor of Philosophy The University of Edinburgh 2019 Declaration I hereby declare that this thesis has been composed by myself and that the work presented within has not been submitted for any other degree or professional qualification. I confirm that the work submitted is my own, except where work which has formed part of jointly-authored publications has been included. My contribution and those of the other authors to this work are indicated below. I confirm that appropriate credit has been given within this thesis where reference has been made to the work of others. I composed this thesis under guidance of Dr. Pippa Thomson. Chapter 2 has been published in PLOS ONE and is attached in the Appendix A, chapter 4 and chapter 5 are published in Translational Psychiatry and are attached in the Appendix C and D, and I expect to submit chapter 6 as a manuscript for publication. -

Systematic Identification and Characterization of Regulatory Elements Derived from Human Endogenous Retroviruses
RESEARCH ARTICLE Systematic identification and characterization of regulatory elements derived from human endogenous retroviruses Jumpei Ito1,2, Ryota Sugimoto1, Hirofumi Nakaoka1,2, Shiro Yamada3, Tetsuaki Kimura1, Takahide Hayano1, Ituro Inoue1,2* 1 Division of Human Genetics, Department of Integrated Genetics, National Institute of Genetics, 1111 Yata, Mishima, Shizuoka, Japan, 2 Department of Genetics, School of Life Science, SOKENDAI (The Graduate a1111111111 University for Advanced Studies), 1111 Yata, Mishima, Shizuoka, Japan, 3 Department of Pediatrics, Tokai a1111111111 University School of Medicine, 143 Shimokasuya, Isehara, Kanagawa, Japan a1111111111 a1111111111 * [email protected] a1111111111 Abstract Human endogenous retroviruses (HERVs) and other long terminal repeat (LTR)-type OPEN ACCESS retrotransposons (HERV/LTRs) have regulatory elements that possibly influence the tran- Citation: Ito J, Sugimoto R, Nakaoka H, Yamada S, scription of host genes. We systematically identified and characterized these regulatory ele- Kimura T, Hayano T, et al. (2017) Systematic ments based on publicly available datasets of ChIP-Seq of 97 transcription factors (TFs) identification and characterization of regulatory provided by ENCODE and Roadmap Epigenomics projects. We determined transcription elements derived from human endogenous retroviruses. PLoS Genet 13(7): e1006883. https:// factor-binding sites (TFBSs) using the ChIP-Seq datasets and identified TFBSs observed doi.org/10.1371/journal.pgen.1006883 on HERV/LTR sequences (HERV-TFBSs). Overall, 794,972 HERV-TFBSs were identified. Editor: CeÂdric Feschotte, University of Utah School Subsequently, we identified ªHERV/LTR-shared regulatory element (HSRE),º defined as a of Medicine, UNITED STATES TF-binding motif in HERV-TFBSs, shared within a substantial fraction of a HERV/LTR type. -

Nº Ref Uniprot Proteína Péptidos Identificados Por MS/MS 1 P01024
Document downloaded from http://www.elsevier.es, day 26/09/2021. This copy is for personal use. Any transmission of this document by any media or format is strictly prohibited. Nº Ref Uniprot Proteína Péptidos identificados 1 P01024 CO3_HUMAN Complement C3 OS=Homo sapiens GN=C3 PE=1 SV=2 por 162MS/MS 2 P02751 FINC_HUMAN Fibronectin OS=Homo sapiens GN=FN1 PE=1 SV=4 131 3 P01023 A2MG_HUMAN Alpha-2-macroglobulin OS=Homo sapiens GN=A2M PE=1 SV=3 128 4 P0C0L4 CO4A_HUMAN Complement C4-A OS=Homo sapiens GN=C4A PE=1 SV=1 95 5 P04275 VWF_HUMAN von Willebrand factor OS=Homo sapiens GN=VWF PE=1 SV=4 81 6 P02675 FIBB_HUMAN Fibrinogen beta chain OS=Homo sapiens GN=FGB PE=1 SV=2 78 7 P01031 CO5_HUMAN Complement C5 OS=Homo sapiens GN=C5 PE=1 SV=4 66 8 P02768 ALBU_HUMAN Serum albumin OS=Homo sapiens GN=ALB PE=1 SV=2 66 9 P00450 CERU_HUMAN Ceruloplasmin OS=Homo sapiens GN=CP PE=1 SV=1 64 10 P02671 FIBA_HUMAN Fibrinogen alpha chain OS=Homo sapiens GN=FGA PE=1 SV=2 58 11 P08603 CFAH_HUMAN Complement factor H OS=Homo sapiens GN=CFH PE=1 SV=4 56 12 P02787 TRFE_HUMAN Serotransferrin OS=Homo sapiens GN=TF PE=1 SV=3 54 13 P00747 PLMN_HUMAN Plasminogen OS=Homo sapiens GN=PLG PE=1 SV=2 48 14 P02679 FIBG_HUMAN Fibrinogen gamma chain OS=Homo sapiens GN=FGG PE=1 SV=3 47 15 P01871 IGHM_HUMAN Ig mu chain C region OS=Homo sapiens GN=IGHM PE=1 SV=3 41 16 P04003 C4BPA_HUMAN C4b-binding protein alpha chain OS=Homo sapiens GN=C4BPA PE=1 SV=2 37 17 Q9Y6R7 FCGBP_HUMAN IgGFc-binding protein OS=Homo sapiens GN=FCGBP PE=1 SV=3 30 18 O43866 CD5L_HUMAN CD5 antigen-like OS=Homo -

Overexpression of the Proneural Transcription Factor ASCL1 In
Malli et al. Molecular Cytogenetics (2018) 11:3 DOI 10.1186/s13039-018-0355-7 CASE REPORT Open Access Overexpression of the proneural transcription factor ASCL1 in chronic lymphocytic leukemia with a t(12;14)(q23.2;q32.3) Theodora Malli1, Melanie Rammer1, Sabrina Haslinger1, Jonathan Burghofer1, Sonja Burgstaller2, Hans-Christian Boesmueller3,4, Renate Marschon1, Wolfgang Kranewitter1, Martin Erdel1, Sabine Deutschbauer1 and Gerald Webersinke1* Abstract Background: Translocations of the IGH locus on 14q32.3 are present in about 8% of patients with chronic lymphocytic leukemia (CLL) and contribute to leukemogenesis by deregulating the expression of the IGH-partner genes. Identification of these genes and investigation of the downstream effects of their deregulation can reveal disease-causing mechanisms. Case presentation: We report on the molecular characterization of a novel t(12;14)(q23.2;q32.3) in CLL. As a consequence of the rearrangement ASCL1 was brought into proximity of the IGHJ-Cμ enhancer and was highly overexpressed in the aberrant B-cells of the patient, as shown by qPCR and immunohistochemistry. ASCL1 encodes for a transcription factor acting as a master regulator of neurogenesis, is overexpressed in neuroendocrine tumors and a promising therapeutic target in small cell lung cancer (SCLC). Its overexpression has also been recently reported in acute adult T-cell leukemia/lymphoma. To examine possible downstream effects of the ASCL1 upregulation in CLL, we compared the gene expression of sorted CD5+ cells of the translocation patient to that of CD19+ B-cells from seven healthy donors and detected 176 significantly deregulated genes (Fold Change ≥2, FDR p ≤ 0.01). -

The Effect of Prpsc Accumulation on Inflammatory Gene
Edinburgh Research Explorer The effect of PrP(Sc) accumulation on inflammatory gene expression within sheep peripheral lymphoid tissue Citation for published version: Gossner, AG & Hopkins, J 2015, 'The effect of PrP(Sc) accumulation on inflammatory gene expression within sheep peripheral lymphoid tissue', Veterinary Microbiology, vol. 181, no. 3-4, pp. 204-211. https://doi.org/10.1016/j.vetmic.2015.10.013 Digital Object Identifier (DOI): 10.1016/j.vetmic.2015.10.013 Link: Link to publication record in Edinburgh Research Explorer Document Version: Publisher's PDF, also known as Version of record Published In: Veterinary Microbiology General rights Copyright for the publications made accessible via the Edinburgh Research Explorer is retained by the author(s) and / or other copyright owners and it is a condition of accessing these publications that users recognise and abide by the legal requirements associated with these rights. Take down policy The University of Edinburgh has made every reasonable effort to ensure that Edinburgh Research Explorer content complies with UK legislation. If you believe that the public display of this file breaches copyright please contact [email protected] providing details, and we will remove access to the work immediately and investigate your claim. Download date: 29. Sep. 2021 G Model VETMIC 7118 No. of Pages 8 Veterinary Microbiology xxx (2015) xxx–xxx Contents lists available at ScienceDirect Veterinary Microbiology journal homepage: www.elsevier.com/locate/vetmic Sc The effect of PrP accumulation on inflammatory gene expression within sheep peripheral lymphoid tissue Anton G. Gossner, John Hopkins* The Roslin Institute & R(D)SVS, University of Edinburgh, Easter Bush, Midlothian EH25 9RG, UK A R T I C L E I N F O A B S T R A C T Sc Article history: Accumulation of the misfolded prion protein, PrP in the central nervous system (CNS) is strongly linked Received 27 April 2015 to progressive neurodegenerative disease. -

Engineered Type 1 Regulatory T Cells Designed for Clinical Use Kill Primary
ARTICLE Acute Myeloid Leukemia Engineered type 1 regulatory T cells designed Ferrata Storti Foundation for clinical use kill primary pediatric acute myeloid leukemia cells Brandon Cieniewicz,1* Molly Javier Uyeda,1,2* Ping (Pauline) Chen,1 Ece Canan Sayitoglu,1 Jeffrey Mao-Hwa Liu,1 Grazia Andolfi,3 Katharine Greenthal,1 Alice Bertaina,1,4 Silvia Gregori,3 Rosa Bacchetta,1,4 Norman James Lacayo,1 Alma-Martina Cepika1,4# and Maria Grazia Roncarolo1,2,4# Haematologica 2021 Volume 106(10):2588-2597 1Department of Pediatrics, Division of Stem Cell Transplantation and Regenerative Medicine, Stanford School of Medicine, Stanford, CA, USA; 2Stanford Institute for Stem Cell Biology and Regenerative Medicine, Stanford School of Medicine, Stanford, CA, USA; 3San Raffaele Telethon Institute for Gene Therapy, Milan, Italy and 4Center for Definitive and Curative Medicine, Stanford School of Medicine, Stanford, CA, USA *BC and MJU contributed equally as co-first authors #AMC and MGR contributed equally as co-senior authors ABSTRACT ype 1 regulatory (Tr1) T cells induced by enforced expression of interleukin-10 (LV-10) are being developed as a novel treatment for Tchemotherapy-resistant myeloid leukemias. In vivo, LV-10 cells do not cause graft-versus-host disease while mediating graft-versus-leukemia effect against adult acute myeloid leukemia (AML). Since pediatric AML (pAML) and adult AML are different on a genetic and epigenetic level, we investigate herein whether LV-10 cells also efficiently kill pAML cells. We show that the majority of primary pAML are killed by LV-10 cells, with different levels of sensitivity to killing. Transcriptionally, pAML sensitive to LV-10 killing expressed a myeloid maturation signature. -
View a Copy of This Licence, Visit
Robertson et al. BMC Biology (2020) 18:103 https://doi.org/10.1186/s12915-020-00826-z RESEARCH ARTICLE Open Access Large-scale discovery of male reproductive tract-specific genes through analysis of RNA-seq datasets Matthew J. Robertson1,2, Katarzyna Kent3,4,5, Nathan Tharp3,4,5, Kaori Nozawa3,5, Laura Dean3,4,5, Michelle Mathew3,4,5, Sandra L. Grimm2,6, Zhifeng Yu3,5, Christine Légaré7,8, Yoshitaka Fujihara3,5,9,10, Masahito Ikawa9, Robert Sullivan7,8, Cristian Coarfa1,2,6*, Martin M. Matzuk1,3,5,6 and Thomas X. Garcia3,4,5* Abstract Background: The development of a safe, effective, reversible, non-hormonal contraceptive method for men has been an ongoing effort for the past few decades. However, despite significant progress on elucidating the function of key proteins involved in reproduction, understanding male reproductive physiology is limited by incomplete information on the genes expressed in reproductive tissues, and no contraceptive targets have so far reached clinical trials. To advance product development, further identification of novel reproductive tract-specific genes leading to potentially druggable protein targets is imperative. Results: In this study, we expand on previous single tissue, single species studies by integrating analysis of publicly available human and mouse RNA-seq datasets whose initial published purpose was not focused on identifying male reproductive tract-specific targets. We also incorporate analysis of additional newly acquired human and mouse testis and epididymis samples to increase the number of targets identified. We detected a combined total of 1178 genes for which no previous evidence of male reproductive tract-specific expression was annotated, many of which are potentially druggable targets. -
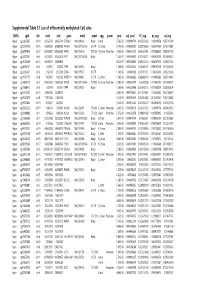
Supplementary Table S1 Kim Et Al.Xlsx
Supplemental Table S1. List of differentially methylated CpG sites DMRs cgid chr start end gene nmid annot cpg prom pval adj. pval FC_avg nc_avg cd_avg Hyper cg12827637 chr14 69256790 69256791 ZFP36L1 NM_004926 Body Island . 5.76E-05 0.000989714 4.465355492 0.06419878 0.287016909 Hyper cg02072495 chr15 60689284 60689285 ANXA2 NM_001136015 5'UTR N_Shore . 2.04E-05 0.000560282 3.808196653 0.056572464 0.215719887 Hyper cg26147845 chr12 132433837 132433838 EP400 NM_015409 TSS1500N_Shore Promoter 3.92E-06 0.000301217 3.643351981 0.077868629 0.283967159 Hyper cg23737061 chr3 60226327 60226328 FHIT NM_001166243 Body . 1.65E-07 0.000239667 3.616168212 0.093920309 0.339893251 Hyper cg18215449 chr12 66089472 66089473 . 4.22E-07 0.000239667 3.565225234 0.064067475 0.228671502 Hyper cg24082121 chr5 672871 672872 TPPP NM_007030 Body . 1.78E-05 0.000525064 3.505843071 0.089095718 0.312606189 Hyper cg23032421 chr3 3152037 3152038 IL5RA NM_175727 5'UTR . 1.04E-05 0.00041338 3.371418731 0.114213604 0.385299026 Hyper cg11737172 chr8 1923051 1923052 KBTBD11 NM_014867 5'UTR S_Shore . 1.38E-05 0.000466865 3.368483202 0.108495063 0.365700645 Hyper cg16886175 chr7 114562062 114562063 MDFIC NM_001166345 TSS200 N_Shore Promoter 7.28E-07 0.000243374 3.35638256 0.101463003 0.340784291 Hyper cg10986412 chr5 672909 672910 TPPP NM_007030 Body . 1.56E-06 0.000255546 3.316242102 0.071981379 0.238939304 Hyper cg02025737 chr15 33384750 33384751 . 3.80E-05 0.000776402 3.312390381 0.13643852 0.452168879 Hyper cg06243675 chr8 11801364 11801365 . 5.23E-05 0.000932639 3.254014485 0.016251643 0.053108482 Hyper cg09577804 chr10 3235531 3235532 . 5.04E-05 0.000912242 3.247042327 0.043496295 0.141459016 Hyper cg23057220 chr19 1356314 1356315 MUM1 NR_024247 TSS200 S_Shore Promoter 3.65E-05 0.000758178 3.204370027 0.14549078 0.466426732 Hyper cg06984883 chr1 1243563 1243564 PUSL1 NM_153339 TSS1500Island Promoter 5.07E-06 0.000323878 3.198831858 0.005047892 0.01636724 Hyper cg15963463 chr1 25253236 25253237 RUNX3 NM_001031680 Body N_Shelf .