Gut Microbiota and the Quality of Oral Anticoagulation in Vitamin K Antagonists Users: a Review of Potential Implications
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

Supplementary Table 2
Supplementary Table 2. Differentially Expressed Genes following Sham treatment relative to Untreated Controls Fold Change Accession Name Symbol 3 h 12 h NM_013121 CD28 antigen Cd28 12.82 BG665360 FMS-like tyrosine kinase 1 Flt1 9.63 NM_012701 Adrenergic receptor, beta 1 Adrb1 8.24 0.46 U20796 Nuclear receptor subfamily 1, group D, member 2 Nr1d2 7.22 NM_017116 Calpain 2 Capn2 6.41 BE097282 Guanine nucleotide binding protein, alpha 12 Gna12 6.21 NM_053328 Basic helix-loop-helix domain containing, class B2 Bhlhb2 5.79 NM_053831 Guanylate cyclase 2f Gucy2f 5.71 AW251703 Tumor necrosis factor receptor superfamily, member 12a Tnfrsf12a 5.57 NM_021691 Twist homolog 2 (Drosophila) Twist2 5.42 NM_133550 Fc receptor, IgE, low affinity II, alpha polypeptide Fcer2a 4.93 NM_031120 Signal sequence receptor, gamma Ssr3 4.84 NM_053544 Secreted frizzled-related protein 4 Sfrp4 4.73 NM_053910 Pleckstrin homology, Sec7 and coiled/coil domains 1 Pscd1 4.69 BE113233 Suppressor of cytokine signaling 2 Socs2 4.68 NM_053949 Potassium voltage-gated channel, subfamily H (eag- Kcnh2 4.60 related), member 2 NM_017305 Glutamate cysteine ligase, modifier subunit Gclm 4.59 NM_017309 Protein phospatase 3, regulatory subunit B, alpha Ppp3r1 4.54 isoform,type 1 NM_012765 5-hydroxytryptamine (serotonin) receptor 2C Htr2c 4.46 NM_017218 V-erb-b2 erythroblastic leukemia viral oncogene homolog Erbb3 4.42 3 (avian) AW918369 Zinc finger protein 191 Zfp191 4.38 NM_031034 Guanine nucleotide binding protein, alpha 12 Gna12 4.38 NM_017020 Interleukin 6 receptor Il6r 4.37 AJ002942 -

Supplemental Table 7. Every Significant Association
Supplemental Table 7. Every significant association between an individual covariate and functional group (assigned to the KO level) as determined by CPGLM regression analysis. Variable Unit RelationshipLabel See also CBCL Aggressive Behavior K05914 + CBCL Emotionally Reactive K05914 + CBCL Externalizing Behavior K05914 + K15665 K15658 CBCL Total K05914 + K15660 K16130 KO: E1.13.12.7; photinus-luciferin 4-monooxygenase (ATP-hydrolysing) [EC:1.13.12.7] :: PFAMS: AMP-binding enzyme; CBQ Inhibitory Control K05914 - K12239 K16120 Condensation domain; Methyltransferase domain; Thioesterase domain; AMP-binding enzyme C-terminal domain LEC Family Separation/Social Services K05914 + K16129 K16416 LEC Poverty Related Events K05914 + K16124 LEC Total K05914 + LEC Turmoil K05914 + CBCL Aggressive Behavior K15665 + CBCL Anxious Depressed K15665 + CBCL Emotionally Reactive K15665 + K05914 K15658 CBCL Externalizing Behavior K15665 + K15660 K16130 KO: K15665, ppsB, fenD; fengycin family lipopeptide synthetase B :: PFAMS: Condensation domain; AMP-binding enzyme; CBCL Total K15665 + K12239 K16120 Phosphopantetheine attachment site; AMP-binding enzyme C-terminal domain; Transferase family CBQ Inhibitory Control K15665 - K16129 K16416 LEC Poverty Related Events K15665 + K16124 LEC Total K15665 + LEC Turmoil K15665 + CBCL Aggressive Behavior K11903 + CBCL Anxiety Problems K11903 + CBCL Anxious Depressed K11903 + CBCL Depressive Problems K11903 + LEC Turmoil K11903 + MODS: Type VI secretion system K01220 K01058 CBCL Anxiety Problems K11906 + CBCL Depressive -

View This Section Focuses on the Genomic and Proteomic Analyses That Were Performed on Methanolobus Vulcani B1d
MIAMI UNIVERSITY The Graduate School Certificate for Approving the Dissertation We hereby approve the Dissertation of Adam John Creighbaum Candidate for the Degree Doctor of Philosophy ______________________________________ Dr. Donald J. Ferguson Jr, Director ______________________________________ Dr. Annette Bollmann, Reader ______________________________________ Dr. Xin Wang, Reader ______________________________________ Dr. Rachael Morgan-Kiss ______________________________________ Dr. Richard Page, Graduate School Representative ABSTRACT EXAMINATION AND RECONSTITUTION OF THE GLYCINE BETAINE- DEPENDENT METHANOGENESIS PATHWAY FROM THE OBLIGATE METHYLOTROPHIC METHANOGEN METHANOLOBUS VULCANI B1D by Adam J. Creighbaum Recent studies indicate that environmentally abundant quaternary amines (QAs) are a primary source for methanogenesis, yet the catabolic enzymes are unknown. We hypothesized that the methanogenic archaeon Methanolobus vulcani B1d metabolizes glycine betaine through a corrinoid-dependent glycine betaine:coenzyme M (CoM) methyl transfer pathway. The draft genome sequence of M. vulcani B1d revealed a gene encoding a predicted non- pyrrolysine MttB homolog (MV8460) with high sequence similarity to the glycine betaine methyltransferase encoded by Desulfitobacterium hafniense Y51. MV8460 catalyzes glycine betaine-dependent methylation of free cob(I)alamin indicating it is an authentic MtgB enzyme. Proteomic analysis revealed that MV8460 and a corrinoid binding protein (MV8465) were highly abundant when M. vulcani B1d was grown -

Structure-Guided Function Discovery of an NRPS-Like Glycine Betaine Reductase for Choline Biosynthesis in Fungi
Structure-guided function discovery of an NRPS-like glycine betaine reductase for choline biosynthesis in fungi Yang Hai (海洋)a, Arthur M. Huangb, and Yi Tanga,b,1 aDepartment of Chemical and Biomolecular Engineering, University of California, Los Angeles, CA 90095; and bDepartment of Chemistry and Biochemistry, University of California, Los Angeles, CA 90095 Edited by Wilfred A. van der Donk, Howard Hughes Medical Institute and University of Illinois, Urbana–Champaign, Urbana, IL, and accepted by Editorial Board Member Stephen J. Benkovic April 10, 2019 (received for review February 27, 2019) Nonribosomal peptide synthetases (NRPSs) and NRPS-like enzymes focused on a fungal CAR-like protein with an unknown function. It have diverse functions in primary and secondary metabolisms. By is distinguished from all other CARs due to an extra C-terminal using a structure-guided approach, we uncovered the function of a YdfG-like short-chain dehydrogenase/reductase domain. We NRPS-like enzyme with unusual domain architecture, catalyzing named this protein ATRR and the corresponding gene atrr after its two sequential two-electron reductions of glycine betaine to choline. unusual domain architecture (A-T-R1-R2). The presence of two Structural analysis based on the homology model suggests cation-π fused R domains in ATRR implies that it could catalyze two con- interactions as the major substrate specificity determinant, which was secutive two-electron reductions of a carboxylic acid to yield an verified using substrate analogs and inhibitors. Bioinformatic analysis alcohol (Fig. 1B). Moreover, a genomic survey reveals that atrr indicates this NRPS-like glycine betaine reductase is highly conserved genes are widespread but exclusive to eukaryotes, mostly in fungi and widespread in kingdom fungi. -

Mediated Host-Microbiome Metabolic Axis Implicated in Health and Disease
1521-009X/44/11/1839–1850$25.00 http://dx.doi.org/10.1124/dmd.116.070615 DRUG METABOLISM AND DISPOSITION Drug Metab Dispos 44:1839–1850, November 2016 Copyright ª 2016 The Author(s) This is an open access article distributed under the CC BY Attribution 4.0 International license. Minireview Trimethylamine and Trimethylamine N-Oxide, a Flavin-Containing Monooxygenase 3 (FMO3)-Mediated Host-Microbiome Metabolic Axis Implicated in Health and Disease Diede Fennema, Ian R. Phillips, and Elizabeth A. Shephard Institute of Structural and Molecular Biology, University College London (D.F., I.R.P., E.A.S.), and School of Biological and Chemical Sciences, Queen Mary University of London (I.R.P.), London, United Kingdom Received March 22, 2016; accepted May 13, 2016 Downloaded from ABSTRACT Flavin-containing monooxygenase 3 (FMO3) is known primarily as an disease, reverse cholesterol transport, and glucose and lipid enzyme involved in the metabolism of therapeutic drugs. On a daily homeostasis. In this review, we consider the dietary components basis, however, we are exposed to one of the most abundant that can give rise to TMA, the gut bacteria involved in the production substrates of the enzyme trimethylamine (TMA), which is released of TMA from dietary precursors, the metabolic reactions by which dmd.aspetjournals.org from various dietary components by the action of gut bacteria. FMO3 bacteria produce and use TMA, and the enzymes that catalyze the converts the odorous TMA to nonodorous TMA N-oxide (TMAO), reactions. Also included is information on bacteria that produce which is excreted in urine. Impaired FMO3 activity gives rise to the TMA in the oral cavity and vagina, two key microbiome niches that inherited disorder primary trimethylaminuria (TMAU). -

Discovery of Industrially Relevant Oxidoreductases
DISCOVERY OF INDUSTRIALLY RELEVANT OXIDOREDUCTASES Thesis Submitted for the Degree of Master of Science by Kezia Rajan, B.Sc. Supervised by Dr. Ciaran Fagan School of Biotechnology Dublin City University Ireland Dr. Andrew Dowd MBio Monaghan Ireland January 2020 Declaration I hereby certify that this material, which I now submit for assessment on the programme of study leading to the award of Master of Science, is entirely my own work, and that I have exercised reasonable care to ensure that the work is original, and does not to the best of my knowledge breach any law of copyright, and has not been taken from the work of others save and to the extent that such work has been cited and acknowledged within the text of my work. Signed: ID No.: 17212904 Kezia Rajan Date: 03rd January 2020 Acknowledgements I would like to thank the following: God, for sending me angels in the form of wonderful human beings over the last two years to help me with any- and everything related to my project. Dr. Ciaran Fagan and Dr. Andrew Dowd, for guiding me and always going out of their way to help me. Thank you for your patience, your advice, and thank you for constantly believing in me. I feel extremely privileged to have gotten an opportunity to work alongside both of you. Everything I’ve learnt and the passion for research that this project has sparked in me, I owe it all to you both. Although I know that words will never be enough to express my gratitude, I still want to say a huge thank you from the bottom of my heart. -

Structure-Guided Function Discovery of an NRPS-Like Glycine Beta
bioRxiv preprint doi: https://doi.org/10.1101/559534; this version posted February 24, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Classification BIOLOGICAL SCIENCES: Biochemistry Structure-guided function discovery of an NRPS-like glycine beta- ine reductase for choline biosynthesis in fungi Yang Hai1, Arthur Huang2, Yi Tang1,2* 1Department of Chemical and Biomolecular Engineering, 2Department of Chemistry and Bio- chemistry, University of California, Los Angeles, California 90095, United States * Email: [email protected] 1 bioRxiv preprint doi: https://doi.org/10.1101/559534; this version posted February 24, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. ABSTRACT Nonribosomal peptide synthetases (NRPS) and NRPS-like enzymes have diverse func- tions in primary and secondary metabolism. By using a structure-guided approach, we uncov- ered the function of an NRPS-like enzyme with unusual domain architecture, catalyzing two sequential two-electron reductions of glycine betaine to choline. Structural analysis based on homology model suggests cation-p interactions as the major substrate specificity determinant, which was verified using substrate analogs and inhibitors. Bioinformatic analysis indicates this NRPS-like glycine betaine reductase is highly conserved and widespread in fungi kingdom. Ge- netic knockout experiments confirmed its role in choline biosynthesis and maintaining glycine betaine homeostasis in fungi. Our findings demonstrate that the oxidative choline-glycine beta- ine degradation pathway can operate in a fully reversible fashion and provide new insights in understanding fungal choline metabolism. -

Mcfadden and Myers (Manuscript)
Trimethylamine N-oxide in Humans and Dairy Cows: Should We Be Concerned? J. W. McFadden and W. A. Myers Department of Animal Science Cornell University Introduction The Greenland shark is an oceanic enigma. The creature holds the recognition as being the longest-living vertebrate in the world. They sexually mature after 100 years and survive for over four centuries. The shark also contains some of the highest biological observed tissue concentrations of a metabolite called trimethylamine N-oxide (TMAO). Although poisonous when consumed fresh, Greenland shark is compressed and dried to lower the TMAO content and produce a fermented and odorous food called hákarl. These ancient ‘shark bites’ are unique but it is the TMAO that has garnered the recent attention of the scientific community. This is because TMAO has been labeled “The New Red-Meat Risk” for heart disease (Abbasi, 2019). Indeed, numerous studies have been published linking higher circulating TMAO concentrations with cardiovascular disease but also non- alcoholic fatty liver disease (NAFLD) in humans (Li et al., 2017b, Roncal et al., 2019, Tan et al., 2019); however, the science is contentious and subject to major criticism.Rese arch has centered on the ability of dietary L -carnitine, choline, or betaine found in red meat, dairy products, chicken, eggs, and fish to be broken down to trimethylamine (TMA) in the gut, which is absorbed and converted to TMAO in liver by the enzyme flavin -containing monooxygenase-3 (FMO3). For the dairy industry, the story of TMAO has several implications. First, increases in endogenous TMAO may indirectly reflect the gastrointestinal degradation and limited bioavailability of choline, betaine, or L -carnitine, which are often fed as rumen-protected supplements to dairy cattle. -
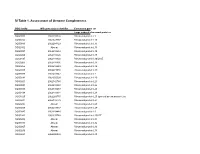
SI Table 1. Assessment of Genome Completeness
SI Table 1. Assessment of Genome Completeness COG family IMG gene object identifier Conserved gene set Large subunit ribosomal proteins COG0081 2062288324 Ribosomal protein L1 COG0244 2062347387 Ribosomal protein L10 COG0080 2062288323 Ribosomal protein L11 COG0102 Absent Ribosomal protein L13 COG0093 2062418832 Ribosomal protein L14 COG0200 2062418826 Ribosomal protein L15 COG0197 2062418838 Ribosomal protein L16/L10E COG0203 2062418836 Ribosomal protein L17 COG0256 2062418829 Ribosomal protein L18 COG0335 2062273558 Ribosomal protein L19 COG0090 2062418842 Ribosomal protein L2 COG0292 2062350539 Ribosomal protein L20 COG0261 2062142780 Ribosomal protein L21 COG0091 2062418840 Ribosomal protein L22 COG0089 2062138283 Ribosomal protein L23 COG0198 2062418834 Ribosomal protein L24 COG1825 2062269715 Ribosomal protein L25 (general stress protein Ctc) COG0211 2062142779 Ribosomal protein L27 COG0227 Absent Ribosomal protein L28 COG0255 2062418837 Ribosomal protein L29 COG0087 2062154483 Ribosomal protein L3 COG1841 2062335748 Ribosomal protein L30/L7E COG0254 Absent Ribosomal protein L31 COG0333 Absent Ribosomal protein L32 COG0267 Absent Ribosomal protein L33 COG0230 Absent Ribosomal protein L34 COG0291 2062350538 Ribosomal protein L35 COG0257 Absent Ribosomal protein L36 COG0088 2062138282 Ribosomal protein L4 COG0094 2062418833 Ribosomal protein L5 COG0097 2062418830 Ribosomal protein L6P/L9E COG0222 2062288326 Ribosomal protein L7/L12 COG0359 2062209880 Ribosomal protein L9 Small subunit ribosomal proteins COG0539 Absent Ribosomal protein -

Investigating Selenium Metabolism in Bacillus Selenitireducens MLS10
WELLS, MICHAEL BANNER, M.S. Se-ing Bacteria in a New Light: Investigating Selenium Metabolism in Bacillus selenitireducens MLS10. (2015) Directed by Dr. Malcolm Schug 84 pp. Prokaryotes metabolize selenium through incorporation into the 21st amino acid selenocysteine, and the respiration of the selenium oxyanions, selenite and selenite.I investigated the physiological function and evolution of selenoproteins inBacillus selenitireducens MLS10 by annotating the selenoproteome of MLS10 and constructing phylogenies of selenoproteins. I investigated the physiology of selenite respiration in MLS10 by obtaining protein profiles using SDS-PAGE, determining the cytochrome content using the pyridine hemochrome assay, and testing for enzyme activity in native gels using selenite-grown MLS10 cells. My research demonstrates that the Bacilli exploit Sec residues far more than has heretofore been appreciated, that the use of Sec residues in MLS10 is ancestral, and suggests that extensive horizontal gene transfer characterizes the evolution of selenoproteins in Gram-positive bacteria and the δ- Proteobacteria. Finally, my research provides evidence that selenite respiration is an inducible respiratory pathway in MLS10, and suggests future directions for further testing of this hypothesis. SE-ING BACTERIA IN A NEW LIGHT: INVESTIGATING SELENIUM METABOLISM IN BACILLUS SELENITIREDUCENS MLS10 by Michael Banner Wells A Thesis Submitted to the Faculty of The Graduate School at The University of North Carolina at Greensboro in Partial Fulfillment of the Requirements for the Degree Master of Science Greensboro 2015 Approved by __________________________ Committee Chair Tezimi Merve Seven’e ithaf ederim. Çalışmalarım boyunca yoluma ışık olduğunuz ve gerçek bilim insanı olmayı bana öğrettiğiniz için size minnettarım. ii APPROVAL PAGE This thesis written by Michael Banner Wells has been approved by the following committee of the Faculty of The Graduate School at The University of North Carolina at Greensboro. -

Supplementary Table 4: Genes Down- and Up-Regulated After Culture in KSOM Vs VIVO
Supplementary table 4: Genes down- and up-regulated after culture in KSOM vs VIVO Fold change Gene symbol Description Affymetrix number -20.25 Rassf4 Ras association (RalGDS/AF-6) domain family 4 1444009_at -14.06 Bhmt2 betaine-homocysteine methyltransferase 2 1418914_s_at -10.56 Akp5 alkaline phosphatase 5 1450635_at -7.02 Socs3 suppressor of cytokine signaling 3 1455899_x_at -5.76 Nucb2 nucleobindin 2 1418355_at -5.76 Stmn3 stathmin-like 3 1435113_x_at -5.29 Bhmt betaine-homocysteine methyltransferase 1450624_at -5.29 Akr1c21 aldo-keto reductase family 1, member C21 1451030_at -5.06 Bmp4 bone morphogenetic protein 4 1422912_at -4.62 Fgf4 fibroblast growth factor 4 1420085_at -4.62 Cbfa2t1h CBFA2T1 identified gene homolog (human) 1444615_x_at -4.41 Arf7 ADP-ribosylation factor 7 1430234_at -4.20 Tmem64 transmembrane protein 64 1434307_at -4.20 Cpxm1 carboxypeptidase X 1 (M14 family) 1448901_at -4.20 Apcdd1 adenomatosis polyposis coli down-regulated 1 1454822_x_at -4.00 Dmgdh dimethylglycine dehydrogenase precursor 1452311_at -3.61 Morc1 microrchidia 1 1419418_a_at -3.61 Tmem64 transmembrane protein 64 1433735_a_at -3.61 Parvb Parvin, beta (Parvb), mRNA 1438672_at -3.61 Akp5 alkaline phosphatase 5 1450636_s_at -3.42 Cebpa CCAAT/enhancer binding protein (C/EBP), alpha 1418982_at -3.24 --- Transcribed locus 1448021_at -3.06 Snap91 synaptosomal-associated protein 91 1416688_at -2.89 MGI:1919030 myo-inositol 1-phosphate synthase A1 1415977_at -2.89 --- RNA transcript from U17 small nucleolar RNA host gene 1433789_at -2.56 Tspan1 tetraspan 1 1417957_a_at -2.56 4921530G04Rik RIKEN cDNA 4921530G04 gene 1460274_at -2.40 Tcl1 T-cell lymphoma breakpoint 1 1422458_at -2.40 Fasn fatty acid synthase 1423828_at -2.25 Tex19 testis expressed gene 19 1417482_at -2.25 Ube2a ubiquitin-conjugating enzyme E2A, RAD6 homolog (S.