May Help Explain Expression of Multiple Genes in Alzheimer's
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Higd1a Is a Positive Regulator of Cytochrome C Oxidase
Higd1a is a positive regulator of cytochrome c oxidase Takaharu Hayashia,b, Yoshihiro Asanoa,b,1, Yasunori Shintania, Hiroshi Aoyamac, Hidetaka Kiokab, Osamu Tsukamotoa, Masahide Hikitad, Kyoko Shinzawa-Itohd, Kazuaki Takafujie, Shuichiro Higoa,b, Hisakazu Katoa, Satoru Yamazakif, Ken Matsuokab, Atsushi Nakanog, Hiroshi Asanumah, Masanori Asakurag, Tetsuo Minaminob, Yu-ichi Gotoi, Takashi Ogurad, Masafumi Kitakazeg, Issei Komuroj, Yasushi Sakatab, Tomitake Tsukiharad,k, Shinya Yoshikawad, and Seiji Takashimaa,k,1 Departments of aMedical Biochemistry and bCardiovascular Medicine, eCenter for Research Education, and cGraduate School of Pharmaceutical Science, Osaka University Graduate School of Medicine, Suita, Osaka 565-0871, Japan; dDepartment of Life Science, University of Hyogo, 3-2-1 Kouto, Kamigohri, Akoh, Hyogo 678-1297, Japan; kCore Research for Evolutional Science and Technology (CREST), Japan Science and Technology Agency, Kawaguchi, Saitama 332-0012, Japan; Departments of fCell Biology and gClinical Research and Development, National Cerebral and Cardiovascular Center Research Institute, Suita, Osaka 565-8565, Japan; hDepartment of Cardiovascular Science and Technology, Kyoto Prefectural University School of Medicine, Kamigyo-ku, Kyoto 602-8566, Japan; iDepartment of Child Neurology, National Center Hospital of Neurology and Psychiatry, National Center of Neurology and Psychiatry, Kodaira, Tokyo 187-8502, Japan; and jDepartment of Cardiovascular Medicine, Graduate School of Medicine, University of Tokyo, Tokyo 113-8656, Japan Edited by Gottfried Schatz, University of Basel, Reinach, Switzerland, and approved December 16, 2014 (received for review October 15, 2014) Cytochrome c oxidase (CcO) is the only enzyme that uses oxygen as we recently revealed (8). Because CcO is the only enzyme in to produce a proton gradient for ATP production during mitochon- the body that can use oxygen for energy transduction, it has been drial oxidative phosphorylation. -

The Genetic Architecture of Hearing Impairment in Mice: Evidence for Frequency Specific 2 Genetic Determinants
G3: Genes|Genomes|Genetics Early Online, published on September 4, 2015 as doi:10.1534/g3.115.021592 1 The genetic architecture of hearing impairment in mice: evidence for frequency specific 2 genetic determinants. 3 4 Amanda L. Crow1, Jeffrey Ohmen2, Juemei Wang3, Joel Lavinsky3, Jaana Hartiala1, Qingzhong 5 Li4, Xin Li5, Pezhman Salehide 3, Eleazar Eskin6, Calvin Pan7, Aldons J. Lusis7, Hooman 6 Allayee1, Rick A. Friedman3 7 8 1Department of Preventive Medicine and Institute for Genetic Medicine, Keck School of 9 Medicine, University of Southern California, Los Angeles, CA 90033 10 2House Ear Institute, Los Angeles, CA 90057 11 3Department of Otolaryngology and Zilkha Neurogenetic Institute, Keck School of Medicine, 12 University of Southern California, Los Angeles, CA, 90033 13 4Department of Otolaryngology - Head and Neck Surgery, Eye & ENT Hospital of Fudan 14 University, Shanghai 200031, China 15 5Clinical Laboratory Department, First Affiliated Hospital of Nanchang University, Nanchang, 16 Jiangxi Province 330006, China 17 6Department of Computer Science and Inter-Departmental Program in Bioinformatics, 18 University of California, Los Angeles, Los Angeles, CA 90095 19 7Departments of Human Genetics, Medicine, and Microbiology, Immunology, and Molecular 20 Genetics, David Geffen School of Medicine at UCLA, Los Angeles, CA 90095 21 © The Author(s) 2013. Published by the Genetics Society of America. 1 Short Title 2 Genetics of Hearing in Mice 3 4 Keywords 5 Genome-wide association study (GWAS), Hybrid Mouse Diversity Panel (HMDP), genetics, 6 genomics, ABR, hearing, cochlear function 7 8 9 Corresponding Author: 10 Rick A. Friedman 11 USC Keck School of Medicine 12 Zilkha Neurogenetic Institute 13 1501 San Pablo Street (ZNI 231) 14 Los Angeles, CA 90033 15 Tel: (323) 442-4843 16 Fax: (323) 442-2059 17 Email: [email protected] 18 19 1 Abstract 2 Genome-wide association studies (GWAS) have been successfully applied in humans for 3 the study of many complex phenotypes. -

Supporting Information
Supporting Information Boothby et al. 10.1073/pnas.1510461112 SI Text tardigrades were allowed to settle and were left overnight at Repetitive Sequences in the H. dujardini Genome. A total of 4,440 room temperature. (10.72%) of the PacBio contigs, making up 7.91% of the as- sembled sequence, do not have direct homologous matches in the DNA Extraction. Isolated specimens were transferred to 1.5-mL × Illumina assembly. These contigs may represent highly repetitive microcentrifuge tubes and centrifuged at 16,837 g for 2 min to sections of the genome that are difficult to assemble with short pellet animals. Excess liquid was removed, and tubes with pel- Illumina reads, or they may be misassemblies of the PacBio data leted specimens were dipped in liquid nitrogen and simulta- neously homogenized with plastic pestles and then left briefly to due to the inherently high rate of sequencing error (10–15%). To thaw. Freeze/thaw douncing was repeated five times to homog- try to resolve this question, we aligned the Illumina reads used to enize samples. To isolate genomic DNA, homogenized samples create our assembly to all assembled PacBio contigs using Bowtie 2. were processed with Qiagen’s DNeasy Blood and Tissue Kit Alignment of our Illumina assembly to our PacBio assembly showed ’ ∼ (catalog no. 69506) according to the manufacturer s instructions. that the average coverage of the differential PacBio contigs is 66% Following isolation, genomic DNA was ethanol-precipitated. of the average coverage across all PacBio contigs, indicating that these sequences are represented in the raw Illumina data but are Assessment of EST and Core Eukaryotic Genes Mapping Approach likely repetitive, and therefore very difficult to assemble accurately. -

1 1 Structural Basis of Mitochondrial Dysfunction in Response to 2
1 2 Structural Basis of Mitochondrial Dysfunction in Response to 3 Cytochrome c Phosphorylation at position 48 4 5 Blas Moreno-Beltrán1,*, Alejandra Guerra-Castellano1,*, Antonio Díaz-Quintana1, 6 Rebecca Del Conte2, Sofía M. García-Mauriño1, Sofía Díaz-Moreno3, Katiuska 7 González-Arzola1, Carlos Santos-Ocaña4, Adrián Velázquez-Campoy5, Miguel A. De la 8 Rosa1, Paola Turano2 & Irene Díaz-Moreno1,# 9 10 1Instituto de Investigaciones Químicas, cicCartuja, Universidad de Sevilla – CSIC, Avda. 11 Américo Vespucio 49, 41092 Seville (Spain). 12 2Magnetic Resonance Center (CERM) – Department of Chemistry, University of 13 Florence, Via Luigi Sacconi 6, 50019 Sesto Fiorentino, Florence (Italy). 14 3Diamond Light Source Ltd., Harwell Science and Innovation Campus, Didcot, 15 Oxfordshire OX11 0DE (United Kingdom). 16 4Centro Andaluz de Biología del Desarrollo, Universidad Pablo de Olavide – CSIC, and 17 CIBERER Instituto de Salud Carlos III, Carretera de Utrera km. 1, 41013 Seville (Spain). 18 5Institute of Biocomputation and Physics of Complex Systems (BIFI), Joint Unit BIFI- 19 IQFR (CSIC), Universidad de Zaragoza, Mariano Esquillor s/n, 50018 Zaragoza (Spain). 20 21 * These authors contribute equally to this work. 22 23 # Correspondence should be addressed to I. D.-M. ([email protected]) 24 Telephone number: +34 954489513; Fax number: +34 954460065 25 1 26 ABSTRACT 27 Regulation of mitochondrial activity allows cells to adapt to changing conditions and to 28 control oxidative stress, and its dysfunction can lead to hypoxia-dependent pathologies, 29 such as ischemia and cancer. Although cytochrome c phosphorylation—in particular, at 30 tyrosine 48—is a key modulator of mitochondrial signaling, its action and molecular basis 31 remain unknown. -

HIGD2A Is Required for Assembly of the COX3 Module of Human Mitochondrial Complex IV
bioRxiv preprint doi: https://doi.org/10.1101/787721; this version posted December 19, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. HIGD2A is required for assembly of the COX3 module of human mitochondrial complex IV Daniella H. Hock1, Boris Reljic1, Ching-Seng Ang2, Hayley S. Mountford3,4,†, Alison G. Compton3,4, Michael T. Ryan5, David R. Thorburn3,4,6 and David A. Stroud*,1 1Department of Biochemistry and Molecular Biology, Bio21 Molecular Science and Biotechnology Institute, University of Melbourne, Parkville, 3052 Victoria, Australia 2Bio21 Mass Spectrometry and Proteomics Facility, The University of Melbourne, Parkville, Victoria 3010, Australia 3Brain and Mitochondrial Research, Murdoch Children's Research Institute, Royal Children's Hospital, Melbourne, Victoria, Australia 4Department of Paediatrics, University of Melbourne, Melbourne, Victoria, Australia 5Department of Biochemistry and Molecular Biology, Monash Biomedicine Discovery Institute, Monash University, 3800 Melbourne, Australia 6Mitochondrial Laboratory, Victorian Clinical Genetics Services, Royal Children's Hospital, Melbourne, Victoria, Australia †Present address: Department of Biological and Medical Sciences, Faculty of Health Sciences, Oxford Brookes University, Oxford OX3 0BP, UK *Correspondence to David A. Stroud Department of Biochemistry and Molecular Biology Bio21 Molecular Science & Biotechnology Institute and The University of Melbourne 30 Flemington Road, Parkville VIC 3052 Australia Email: [email protected] Telephone: +61 3 83447316 Running title: HIGD2A is critical in assembly of human complex IV 1 bioRxiv preprint doi: https://doi.org/10.1101/787721; this version posted December 19, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. -

Stool-Derived Eukaryotic RNA Biomarkers for Detection of High-Risk Adenomas
bioRxiv preprint doi: https://doi.org/10.1101/534412; this version posted January 29, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. Stool-derived eukaryotic RNA biomarkers for detection of high-risk adenomas Erica Barnell1,2*, Yiming Kang2,3*, Andrew Barnell2, Katie Campbell1,2, Kimberly R. Kruse2, Elizabeth M. Wurtzler2, Malachi Griffith1,4,5,6, Aadel A. Chaudhuri3,6,7+, Obi L. Griffith1,4,5,6+ 1 McDonnell Genome Institute, Washington University School of Medicine, St. Louis, MO, USA 2 Geneoscopy LLC, Division of Gastroenterology and Hepatology, St. Louis, MO, USA 3 Department of Computer Science and Engineering, Washington University, St. Louis, MO, USA 4 Department of Genetics, Washington University School of Medicine, St. Louis, MO, USA 5 Division of Oncology, Department of Medicine, Washington University School of Medicine, St. Louis, MO, USA 6 Siteman Cancer Center, Washington University School of Medicine, St. Louis, MO, USA 7 Department of Radiation Oncology, Washington University School of Medicine, St. Louis, MO, USA +Corresponding author *These authors contributed equally to this work. Abbreviations AUC area under the curve LRA low-risk adenoma ANOVA analysis of variance MRA medium-risk adenoma CRC colorectal cancer NPV negative predictive value DE differentially expressed PPV positive predictive value FC fold-change ROC receiver operating characteristic FIT fecal immunochemical test seRNA stool-derived eukaryotic RNA HRA high-risk adenoma Abstract Background and aims: Colorectal cancer (CRC) is the second leading cause of cancer related deaths in the United States. -

Targeted Sequencing Informs the Evaluation of Normal Karyotype Cytopenic Patients for Low-Grade Myelodysplastic Syndrome
Letters to the Editor 2422 chronic lymphocytic leukemia are mostly derived from independent clones. 13 Binder M, Muller F, Jackst A, Lechenne B, Pantic M, Bacher U et al. B-cell receptor Haematologica 2014; 99: 329–338. epitope recognition correlates with the clinical course of chronic lymphocytic 9 Sanchez ML, Almeida J, Gonzalez D, Gonzalez M, Garcia-Marcos MA, leukemia. Cancer 2011; 117: 1891–1900. Balanzategui A et al. Incidence and clinicobiologic characteristics of leukemic 14 Seiler T, Woelfle M, Yancopoulos S, Catera R, Li W, Hatzi K et al. Characteri- B-cell chronic lymphoproliferative disorders with more than one B-cell clone. zation of structurally defined epitopes recognized by monoclonal antibodies pro- Blood 2003; 102: 2994–3002. duced by chronic lymphocytic leukemia B cells. Blood 2009; 114:3615–3624. 10 Klinger M, Zheng J, Elenitoba-Johnson KS, Perkins SL, Faham M, Bahler DW. Next- generation IgVH sequencing CLL-like monoclonal B-cell lymphocytosis reveals frequent oligoclonality and ongoing hypermutation. Leukemia 2015; 30: This work is licensed under a Creative Commons Attribution- 1055–1061. NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons 11 Tiller T, Meffre E, Yurasov S, Tsuiji M, Nussenzweig MC, Wardemann H. license, unless indicated otherwise in the credit line; if the material is not included under Efficient generation of monoclonal antibodies from single human B cells by single the Creative Commons license, users will need to obtain permission from the license cell RT-PCR and expression vector cloning. J Immunol Methods 2008; 329: holder to reproduce the material. -

Role of Cytochrome C Oxidase Nuclear-Encoded Subunits in Health and Disease
Physiol. Res. 69: 947-965, 2020 https://doi.org/10.33549/physiolres.934446 REVIEW Role of Cytochrome c Oxidase Nuclear-Encoded Subunits in Health and Disease Kristýna ČUNÁTOVÁ1, David PAJUELO REGUERA1, Josef HOUŠTĚK1, Tomáš MRÁČEK1, Petr PECINA1 1Department of Bioenergetics, Institute of Physiology, Czech Academy of Sciences, Prague, Czech Republic Received February 2, 2020 Accepted September 13, 2020 Epub Ahead of Print November 2, 2020 Summary [email protected] and Tomáš Mráček, Department of Cytochrome c oxidase (COX), the terminal enzyme of Bioenergetics, Institute of Physiology CAS, Vídeňská 1083, 142 mitochondrial electron transport chain, couples electron transport 20 Prague 4, Czech Republic. E-mail: [email protected] to oxygen with generation of proton gradient indispensable for the production of vast majority of ATP molecules in mammalian Cytochrome c oxidase cells. The review summarizes current knowledge of COX structure and function of nuclear-encoded COX subunits, which may Energy demands of mammalian cells are mainly modulate enzyme activity according to various conditions. covered by ATP synthesis carried out by oxidative Moreover, some nuclear-encoded subunits possess tissue-specific phosphorylation apparatus (OXPHOS) located in the and development-specific isoforms, possibly enabling fine-tuning central bioenergetic organelle, mitochondria. OXPHOS is of COX function in individual tissues. The importance of nuclear- composed of five multi-subunit complexes embedded in encoded subunits is emphasized by recently discovered the inner mitochondrial membrane (IMM). Electron pathogenic mutations in patients with severe mitopathies. In transport from reduced substrates of complexes I and II to addition, proteins substoichiometrically associated with COX were cytochrome c oxidase (COX, complex IV, CIV) is found to contribute to COX activity regulation and stabilization of achieved by increasing redox potential of individual the respiratory supercomplexes. -

HIG1 Antibody (Pab)
21.10.2014HIG1 antibody (pAb) Rabbit Anti-Human Hypoxia-inducible gene 1 protein (HIGD1A, HSPC010) Instruction Manual Catalog Number PK-AB718-6489 Synonyms HIG1 Antibody: Hypoxia-inducible gene 1 protein, HIGD1A, hypoxia inducible domain family 1A, HSPC010 Description HIG1 and HIG2 (Hypoxia-inducible gene 1 and 2, respectively) are known to be induced by hypoxic conditions. HIG1 is induced by hypoxia and by glucose deprivation in cultured cells. In addition, tumor xenografts derived from human cervical cancer cells display increased expression of HIG1 and HIG2 when they are deprived of oxygen. Unlike HIG2, which is ubiquitously expressed and might be an activator and target of the canonical Wnt pathway, the function and the mechanisms underlying its regulation of HIG1 still remained unknown. The putative link between hypoxia and an oncogenic signaling pathway might play an important role in tumorigenesis. Quantity 100 µg Source / Host Rabbit Immunogen HIG1 antibody was raised against a 19 amino acid synthetic peptide near the amino terminus of human HIG1. Purification Method Affinity chromatography purified via peptide column. Clone / IgG Subtype Polyclonal antibody Species Reactivity Human Specificity HIG1 antibody is predicted to not cross-react with HIG2 Formulation Antibody is supplied in PBS containing 0.02% sodium azide. Reconstitution During shipment, small volumes of antibody will occasionally become entrapped in the seal of the product vial. For products with volumes of 200 μl or less, we recommend gently tapping the vial on a hard surface or briefly centrifuging the vial in a tabletop centrifuge to dislodge any liquid in the container’s cap. Storage & Stability Antibody can be stored at 4ºC for three months and at -20°C for up to one year. -

Influenza-Specific Effector Memory B Cells Predict Long-Lived Antibody Responses to Vaccination in Humans
bioRxiv preprint doi: https://doi.org/10.1101/643973; this version posted February 18, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Influenza-specific effector memory B cells predict long-lived antibody responses to vaccination in humans Anoma Nellore1, Esther Zumaquero2, Christopher D. Scharer3, Rodney G. King2, Christopher M. Tipton4, Christopher F. Fucile5, Tian Mi3, Betty Mousseau2, John E. Bradley6, Fen Zhou2, Paul A. Goepfert1, Jeremy M. Boss3, Troy D. Randall6, Ignacio Sanz4, Alexander F. Rosenberg2,5, Frances E. Lund2 1Dept. of Medicine, Division of Infectious Disease, 2Dept of Microbiology, 5Informatics Institute, 6Dept. of Medicine, Division of Clinical Immunology and Rheumatology and at The University of Alabama at Birmingham, Birmingham, AL 35294 USA 3Dept. of Microbiology and Immunology and 4Department of Medicine, Division of Rheumatology Emory University, Atlanta, GA 30322, USA Correspondence should be addressed to: Frances E. Lund, PhD Charles H. McCauley Professor and Chair Dept of Microbiology University of Alabama at Birmingham 276 BBRB Box 11 1720 2nd Avenue South Birmingham AL 35294-2170 [email protected] SHORT RUNNING TITLE: Effector memory B cell development after influenza vaccination 1 bioRxiv preprint doi: https://doi.org/10.1101/643973; this version posted February 18, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract Seasonal influenza vaccination elicits hemagglutinin (HA)-specific CD27+ memory B cells (Bmem) that differ in expression of T-bet, BACH2 and TCF7. -

Application of RAD-Seq in Evolutionary Genomics of Non-Model Organisms
Application of RAD-seq in Evolutionary Genomics of Non-Model Organisms By Boryana S. Koseva Submitted to the graduate degree program in Ecology and Evolutionary Biology and the Graduate Faculty of the University of Kansas in partial fulfillment of the requirements for the degree of Doctor of Philosophy. Chair: John K. Kelly Stuart J. Macdonald Justin P. Blumenstiel Jamie R. Walters Mark T. Holder Date Defended: 28 April 2017 ii The dissertation committee for Boryana Koseva certifies that this is the approved version of the following dissertation: Application of RAD-seq in Evolutionary Genomics of Non-Model Organisms Chair: John K. Kelly Date Approved: 11 May 2017 iii Abstract Next generation sequencing (NGS) technologies are revolutionizing how we study genetics and evolution in the modern world. Data is generated at such a fast pace that scientists are struggling to keep up with the innovations in methodology and analytical tools. Genomes are being sequenced at an unprecedented rate, and scientists in fields that until recently found no use in learning molecular techniques are venturing into the world of high-throughput sequencing. Almost 10 years ago, a research group developed Restriction-site Associated DNA Sequencing (RAD- seq), a method that targets polymorphisms in close proximity to restriction cut sites in hundreds of samples simultaneously. The beauty of RAD-seq lies in that it is highly customizable and it does not require a reference genome, or intimate prior knowledge of the genetics of the study organism one would like to use. The most exciting part about new RAD-seq methods being developed is that their accessibility has opened the door to many non-model organisms to be used in new areas of research. -
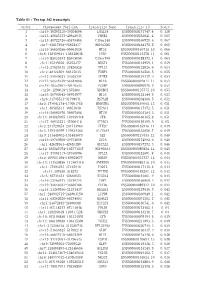
Table S1 - the Top 342 Transcripts
Table S1 - The top 342 transcripts Order Chromosome Position Transcript Name Transcript ID Score 1 chr19:39292310-39304004 LGALS4 ENSG00000171747.4 0.138 2 chr15:40643233-40648635 PHGR1 ENSG00000233041.4 0.085 3 chr15:45722726-45878488 C15orf48 ENSG00000166920.6 0.067 4 chr7:92817898-92855837 HEPACAM2 ENSG00000188175.5 0.065 5 chr16:56659386-56661024 MT1E ENSG00000169715.10 0.066 6 chr6:138409641-138428648 PERP ENSG00000112378.11 0.061 7 chr10:85933493-85945050 C10orf99 ENSG00000188373.4 0.063 8 chr1:45249256-45253377 BEST4 ENSG00000142959.4 0.059 9 chr13:27825445-27830828 RPL21 ENSG00000122026.6 0.054 10 chr2:88422509-88427635 FABP1 ENSG00000163586.5 0.055 11 chr12:39040623-39303394 CPNE8 ENSG00000139117.9 0.054 12 chr12:56544579-56584068 MYL6 ENSG00000092841.14 0.053 13 chr19:40353962-40440533 FCGBP ENSG00000090920.9 0.052 14 chr20:1290618-1373806 SDCBP2 ENSG00000125775.10 0.053 15 chr16:56700642-56701977 MT1G ENSG00000125144.9 0.053 16 chr6:31795511-31798031 HSPA1B ENSG00000204388.5 0.052 17 chr5:179041178-179061785 HNRNPH1 ENSG00000169045.13 0.051 18 chr1:46505811-46651630 TSPAN1 ENSG00000117472.5 0.051 19 chr16:56662970-56667898 MT1M ENSG00000205364.3 0.051 20 chr14:103985995-103989448 CKB ENSG00000166165.8 0.051 21 chr17:46952221-47006418 ATP5G1 ENSG00000159199.9 0.05 22 chr2:231729353-231743963 ITM2C ENSG00000135916.11 0.049 23 chr5:149340299-149432386 SLC26A2 ENSG00000155850.7 0.049 24 chr9:131445702-131458679 SET ENSG00000119335.12 0.048 25 chr16:29789560-29793096 ZG16 ENSG00000174992.6 0.046 26 chr1:42628361-42630389 GUCA2A ENSG00000197273.3