The Hidden Genomic and Transcriptomic Plasticity of Giant Marker Chromosomes in Cancer
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Identification of the Binding Partners for Hspb2 and Cryab Reveals
Brigham Young University BYU ScholarsArchive Theses and Dissertations 2013-12-12 Identification of the Binding arP tners for HspB2 and CryAB Reveals Myofibril and Mitochondrial Protein Interactions and Non- Redundant Roles for Small Heat Shock Proteins Kelsey Murphey Langston Brigham Young University - Provo Follow this and additional works at: https://scholarsarchive.byu.edu/etd Part of the Microbiology Commons BYU ScholarsArchive Citation Langston, Kelsey Murphey, "Identification of the Binding Partners for HspB2 and CryAB Reveals Myofibril and Mitochondrial Protein Interactions and Non-Redundant Roles for Small Heat Shock Proteins" (2013). Theses and Dissertations. 3822. https://scholarsarchive.byu.edu/etd/3822 This Thesis is brought to you for free and open access by BYU ScholarsArchive. It has been accepted for inclusion in Theses and Dissertations by an authorized administrator of BYU ScholarsArchive. For more information, please contact [email protected], [email protected]. Identification of the Binding Partners for HspB2 and CryAB Reveals Myofibril and Mitochondrial Protein Interactions and Non-Redundant Roles for Small Heat Shock Proteins Kelsey Langston A thesis submitted to the faculty of Brigham Young University in partial fulfillment of the requirements for the degree of Master of Science Julianne H. Grose, Chair William R. McCleary Brian Poole Department of Microbiology and Molecular Biology Brigham Young University December 2013 Copyright © 2013 Kelsey Langston All Rights Reserved ABSTRACT Identification of the Binding Partners for HspB2 and CryAB Reveals Myofibril and Mitochondrial Protein Interactors and Non-Redundant Roles for Small Heat Shock Proteins Kelsey Langston Department of Microbiology and Molecular Biology, BYU Master of Science Small Heat Shock Proteins (sHSP) are molecular chaperones that play protective roles in cell survival and have been shown to possess chaperone activity. -

The Endocytic Membrane Trafficking Pathway Plays a Major Role
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by University of Liverpool Repository RESEARCH ARTICLE The Endocytic Membrane Trafficking Pathway Plays a Major Role in the Risk of Parkinson’s Disease Sara Bandres-Ciga, PhD,1,2 Sara Saez-Atienzar, PhD,3 Luis Bonet-Ponce, PhD,4 Kimberley Billingsley, MSc,1,5,6 Dan Vitale, MSc,7 Cornelis Blauwendraat, PhD,1 Jesse Raphael Gibbs, PhD,7 Lasse Pihlstrøm, MD, PhD,8 Ziv Gan-Or, MD, PhD,9,10 The International Parkinson’s Disease Genomics Consortium (IPDGC), Mark R. Cookson, PhD,4 Mike A. Nalls, PhD,1,11 and Andrew B. Singleton, PhD1* 1Molecular Genetics Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Bethesda, Maryland, USA 2Instituto de Investigación Biosanitaria de Granada (ibs.GRANADA), Granada, Spain 3Transgenics Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Bethesda, Maryland, USA 4Cell Biology and Gene Expression Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Bethesda, Maryland, USA 5Department of Molecular and Clinical Pharmacology, Institute of Translational Medicine, University of Liverpool, Liverpool, United Kingdom 6Department of Pathophysiology, University of Tartu, Tartu, Estonia 7Computational Biology Group, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Bethesda, Maryland, USA 8Department of Neurology, Oslo University Hospital, Oslo, Norway 9Department of Neurology and Neurosurgery, Department of Human Genetics, McGill University, Montréal, Quebec, Canada 10Department of Neurology and Neurosurgery, Montreal Neurological Institute, McGill University, Montréal, Quebec, Canada 11Data Tecnica International, Glen Echo, Maryland, USA ABSTRACT studies, summary-data based Mendelian randomization Background: PD is a complex polygenic disorder. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Targeting Hedgehog Signalling Through the Ubiquitylation Process: the Multiple Roles of the HECT-E3 Ligase Itch
cells Review Targeting Hedgehog Signalling through the Ubiquitylation Process: The Multiple Roles of the HECT-E3 Ligase Itch Paola Infante 1,†, Ludovica Lospinoso Severini 2,† , Flavia Bernardi 2, Francesca Bufalieri 2 and Lucia Di Marcotullio 2,3,* 1 Center for Life NanoScience@Sapienza, Istituto Italiano di Tecnologia, 00161 Rome, Italy; [email protected] 2 Department of Molecular Medicine, University of Rome La Sapienza, 00161 Rome, Italy; [email protected] (L.L.S.); fl[email protected] (F.B.); [email protected] (F.B.) 3 Istituto Pasteur-Fondazione Cenci Bolognetti, University of Rome La Sapienza, 00161 Rome, Italy * Correspondence: [email protected]; Tel.: +39-06-49255657 † These authors contributed equally to this work. Received: 28 December 2018; Accepted: 26 January 2019; Published: 29 January 2019 Abstract: Hedgehog signalling (Hh) is a developmental conserved pathway strongly involved in cancers when deregulated. This important pathway is orchestrated by numerous regulators, transduces through distinct routes and is finely tuned at multiple levels. In this regard, ubiquitylation processes stand as essential for controlling Hh pathway output. Although this post-translational modification governs proteins turnover, it is also implicated in non-proteolytic events, thereby regulating the most important cellular functions. The HECT E3 ligase Itch, well known to control immune response, is emerging to have a pivotal role in tumorigenesis. By illustrating Itch specificities on Hh signalling key components, here we review the role of this HECT E3 ubiquitin ligase in suppressing Hh-dependent tumours and explore its potential as promising target for innovative therapeutic approaches. Keywords: Hedgehog; cancer; ubiquitylation; Itch; Numb; β-arrestin2; GLI1; SuFu; Patched1 1. -

The Complex SNP and CNV Genetic Architecture of the Increased Risk of Congenital Heart Defects in Down Syndrome
Downloaded from genome.cshlp.org on September 24, 2021 - Published by Cold Spring Harbor Laboratory Press Research The complex SNP and CNV genetic architecture of the increased risk of congenital heart defects in Down syndrome M. Reza Sailani,1,2 Periklis Makrythanasis,1 Armand Valsesia,3,4,5 Federico A. Santoni,1 Samuel Deutsch,1 Konstantin Popadin,1 Christelle Borel,1 Eugenia Migliavacca,1 Andrew J. Sharp,1,20 Genevieve Duriaux Sail,1 Emilie Falconnet,1 Kelly Rabionet,6,7,8 Clara Serra-Juhe´,7,9 Stefano Vicari,10 Daniela Laux,11 Yann Grattau,12 Guy Dembour,13 Andre Megarbane,12,14 Renaud Touraine,15 Samantha Stora,12 Sofia Kitsiou,16 Helena Fryssira,16 Chariklia Chatzisevastou-Loukidou,16 Emmanouel Kanavakis,16 Giuseppe Merla,17 Damien Bonnet,11 Luis A. Pe´rez-Jurado,7,9 Xavier Estivill,6,7,8 Jean M. Delabar,18 and Stylianos E. Antonarakis1,2,19,21 1–19[Author affiliations appear at the end of the paper.] Congenital heart defect (CHD) occurs in 40% of Down syndrome (DS) cases. While carrying three copies of chromosome 21 increases the risk for CHD, trisomy 21 itself is not sufficient to cause CHD. Thus, additional genetic variation and/or environmental factors could contribute to the CHD risk. Here we report genomic variations that in concert with trisomy 21, determine the risk for CHD in DS. This case-control GWAS includes 187 DS with CHD (AVSD = 69, ASD = 53, VSD = 65) as cases, and 151 DS without CHD as controls. Chromosome 21–specific association studies revealed rs2832616 and rs1943950 as CHD risk alleles (adjusted genotypic P-values <0.05). -

RET Gene Fusions in Malignancies of the Thyroid and Other Tissues
G C A T T A C G G C A T genes Review RET Gene Fusions in Malignancies of the Thyroid and Other Tissues Massimo Santoro 1,*, Marialuisa Moccia 1, Giorgia Federico 1 and Francesca Carlomagno 1,2 1 Department of Molecular Medicine and Medical Biotechnology, University of Naples “Federico II”, 80131 Naples, Italy; [email protected] (M.M.); [email protected] (G.F.); [email protected] (F.C.) 2 Institute of Endocrinology and Experimental Oncology of the CNR, 80131 Naples, Italy * Correspondence: [email protected] Received: 10 March 2020; Accepted: 12 April 2020; Published: 15 April 2020 Abstract: Following the identification of the BCR-ABL1 (Breakpoint Cluster Region-ABelson murine Leukemia) fusion in chronic myelogenous leukemia, gene fusions generating chimeric oncoproteins have been recognized as common genomic structural variations in human malignancies. This is, in particular, a frequent mechanism in the oncogenic conversion of protein kinases. Gene fusion was the first mechanism identified for the oncogenic activation of the receptor tyrosine kinase RET (REarranged during Transfection), initially discovered in papillary thyroid carcinoma (PTC). More recently, the advent of highly sensitive massive parallel (next generation sequencing, NGS) sequencing of tumor DNA or cell-free (cfDNA) circulating tumor DNA, allowed for the detection of RET fusions in many other solid and hematopoietic malignancies. This review summarizes the role of RET fusions in the pathogenesis of human cancer. Keywords: kinase; tyrosine kinase inhibitor; targeted therapy; thyroid cancer 1. The RET Receptor RET (REarranged during Transfection) was initially isolated as a rearranged oncoprotein upon the transfection of a human lymphoma DNA [1]. -
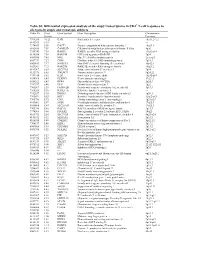
Table S1| Differential Expression Analysis of the Atopy Transcriptome
Table S1| Differential expression analysis of the atopy transcriptome in CD4+ T-cell responses to allergens in atopic and nonatopic subjects Probe ID S.test Gene Symbol Gene Description Chromosome Statistic Location 7994280 10.32 IL4R Interleukin 4 receptor 16p11.2-12.1 8143383 8.95 --- --- --- 7974689 8.50 DACT1 Dapper, antagonist of beta-catenin, homolog 1 14q23.1 8102415 7.59 CAMK2D Calcium/calmodulin-dependent protein kinase II delta 4q26 7950743 7.58 RAB30 RAB30, member RAS oncogene family 11q12-q14 8136580 7.54 RAB19B GTP-binding protein RAB19B 7q34 8043504 7.45 MAL Mal, T-cell differentiation protein 2cen-q13 8087739 7.27 CISH Cytokine inducible SH2-containing protein 3p21.3 8000413 7.17 NSMCE1 Non-SMC element 1 homolog (S. cerevisiae) 16p12.1 8021301 7.15 RAB27B RAB27B, member RAS oncogene family 18q21.2 8143367 6.83 SLC37A3 Solute carrier family 37 member 3 7q34 8152976 6.65 TMEM71 Transmembrane protein 71 8q24.22 7931914 6.56 IL2R Interleukin 2 receptor, alpha 10p15-p14 8014768 6.43 PLXDC1 Plexin domain containing 1 17q21.1 8056222 6.43 DPP4 Dipeptidyl-peptidase 4 (CD26) 2q24.3 7917697 6.40 GFI1 Growth factor independent 1 1p22 7903507 6.39 FAM102B Family with sequence similarity 102, member B 1p13.3 7968236 5.96 RASL11A RAS-like, family 11, member A --- 7912537 5.95 DHRS3 Dehydrogenase/reductase (SDR family) member 3 1p36.1 7963491 5.83 KRT1 Keratin 1 (epidermolytic hyperkeratosis) 12q12-q13 7903786 5.72 CSF1 Colony stimulating factor 1 (macrophage) 1p21-p13 8019061 5.67 SGSH N-sulfoglucosamine sulfohydrolase (sulfamidase) 17q25.3 -

© Copyright 2021 Heather Raquel Dahlin
© Copyright 2021 Heather Raquel Dahlin The Structure of Sperm Autoantigenic Protein (SPA17): An R2D2 Protein Critical to Cilia and Implicated in Oncogenesis Heather Raquel Dahlin A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy University of Washington 2021 Reading Committee: John D. Scott, Chair Ning Zheng Linda Wordeman Program Authorized to Offer Degree: Pharmacology University of Washington Abstract Structure of SPA17: An R2D2 Protein Critical to Cilia and Implicated in Oncogenesis Heather Raquel Dahlin Chair of the Supervisory Committee: John D. Scott, Ph.D., Edwin G. Krebs- Speights Professor of Cell Signaling and Cancer Biology Pharmacology A-Kinase Anchoring proteins (AKAPs) localize the activity of cyclic AMP (cAMP)-Dependent Protein Kinase (PKA) through interaction of an amphipathic helix that binds to a conserved RIIα docking and dimerization (R2D2) domain on the N-terminus of PKA. Genome analysis indicates that at least thirteen other RIIα superfamily proteins exist in humans, which are not coupled to cyclic nucleotide binding domains and are largely localized to cilia and flagella. The newly reported R2D2 proteins exist in two lineages differing by their similarity to Type I or Type II PKA. Moreover, R2D2 domains bind to AKAPs and can contain extra regulatory sequences conferring novel functions and binding specificity. Here we detail the structure of one such domain comprising the N-terminus of Sperm Autoantigenic Protein 17 (SPA17) resolved to 1.72 Å. The structure of core hydrophobic sites for dimerization and AKAP binding are highly conserved between PKA and SPA17. Additional flanking sequences outside of the core R2D2 domain occlude the AKAP binding site and reduce the affinity for AKAP helices in the absence of heterodimerization with another R2D2 protein, ROPN1L. -
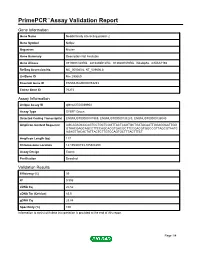
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name Nedd4 family interacting protein 2 Gene Symbol Ndfip2 Organism Mouse Gene Summary Description Not Available Gene Aliases 0710001O20Rik, 2810436B12Rik, 9130207N19Rik, N4wbp5a, mKIAA1165 RefSeq Accession No. NC_000080.6, NT_039606.8 UniGene ID Mm.290669 Ensembl Gene ID ENSMUSG00000053253 Entrez Gene ID 76273 Assay Information Unique Assay ID qMmuCED0039954 Assay Type SYBR® Green Detected Coding Transcript(s) ENSMUST00000181969, ENSMUST00000138283, ENSMUST00000136040 Amplicon Context Sequence AGCAGAGCCCAGTCCTGCTCGGTTACTCAGTGCTGATACAATTGGAGGAATTGG GTAACGAGCAGCCTTCCAGCACGTGACGCTTCCGACGTGGCCGTTAGCGTAATC AGAGTTACACTATTACTCTTGTCCAGTGCTTTACTTTCT Amplicon Length (bp) 117 Chromosome Location 14:105308153-105308299 Assay Design Exonic Purification Desalted Validation Results Efficiency (%) 99 R2 0.996 cDNA Cq 20.52 cDNA Tm (Celsius) 83.5 gDNA Cq 23.84 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 1/4 PrimePCR™Assay Validation Report Ndfip2, Mouse Amplification Plot Amplification of cDNA generated from 25 ng of universal reference RNA Melt Peak Melt curve analysis of above amplification Standard Curve Standard curve generated using 20 million copies of template diluted 10-fold to 20 copies Page 2/4 PrimePCR™Assay Validation Report Products used to generate validation data Real-Time PCR Instrument CFX384 Real-Time PCR Detection System Reverse Transcription Reagent iScript™ Advanced cDNA Synthesis Kit for RT-qPCR Real-Time PCR Supermix SsoAdvanced™ SYBR® Green Supermix Experimental Sample qPCR Mouse Reference Total RNA Data Interpretation Unique Assay ID This is a unique identifier that can be used to identify the assay in the literature and online. Detected Coding Transcript(s) This is a list of the Ensembl transcript ID(s) that this assay will detect. -

CDC123 Antibody (RQ6088)
CDC123 Antibody (RQ6088) Catalog No. Formulation Size RQ6088 0.5mg/ml if reconstituted with 0.2ml sterile DI water 100 ug Bulk quote request Availability 1-3 business days Species Reactivity Human Format Antigen affinity purified Clonality Polyclonal (rabbit origin) Isotype Rabbit IgG Purity Affinity purified Buffer Lyophilized from 1X PBS with 2% Trehalose and 0.025% sodium azide UniProt O75794 Localization Cytoplasmic Applications Western blot : 1-2ug/ml Immunohistochemistry (FFPE) : 2-5ug/ml Immunofluorescence : 5ug/ml Flow cytometry : 1-3ug/million cells Direct ELISA : 0.1-0.5ug/ml Limitations This CDC123 antibody is available for research use only. Immunofluorescent staining of FFPE human A431 cells with CDC123 antibody (green) and DAPI nuclear stain (blue). HIER: steam section in pH6 citrate buffer for 20 min. IHC staining of FFPE human pancreatic cancer with CDC123 antibody. HIER: boil tissue sections in pH8 EDTA for 20 min and allow to cool before testing. IHC staining of FFPE human bladder cancer with CDC123 antibody. HIER: boil tissue sections in pH8 EDTA for 20 min and allow to cool before testing. IHC staining of FFPE human appendicitis tissue with CDC123 antibody. HIER: boil tissue sections in pH8 EDTA for 20 min and allow to cool before testing. IHC staining of FFPE human ovarian cancer with CDC123 antibody. HIER: boil tissue sections in pH8 EDTA for 20 min and allow to cool before testing. IHC staining of FFPE human liver cancer with CDC123 antibody. HIER: boil tissue sections in pH8 EDTA for 20 min and allow to cool before testing. IHC staining of FFPE human gastric cancer with CDC123 antibody. -

Miz1 Is Required to Maintain Autophagic Flux
ARTICLE Received 3 Apr 2013 | Accepted 3 Sep 2013 | Published 3 Oct 2013 DOI: 10.1038/ncomms3535 Miz1 is required to maintain autophagic flux Elmar Wolf1,*, Anneli Gebhardt1,*, Daisuke Kawauchi2, Susanne Walz1, Bjo¨rn von Eyss1, Nicole Wagner3, Christoph Renninger3, Georg Krohne1, Esther Asan3, Martine F. Roussel2 & Martin Eilers1,4 Miz1 is a zinc finger protein that regulates the expression of cell cycle inhibitors as part of a complex with Myc. Cell cycle-independent functions of Miz1 are poorly understood. Here we use a Nestin-Cre transgene to delete an essential domain of Miz1 in the central nervous system (Miz1DPOZNes). Miz1DPOZNes mice display cerebellar neurodegeneration characterized by the progressive loss of Purkinje cells. Chromatin immunoprecipitation sequencing and biochemical analyses show that Miz1 activates transcription upon binding to a non-palin- dromic sequence present in core promoters. Target genes of Miz1 encode regulators of autophagy and proteins involved in vesicular transport that are required for autophagy. Miz1DPOZ neuronal progenitors and fibroblasts show reduced autophagic flux. Consistently, polyubiquitinated proteins and p62/Sqtm1 accumulate in the cerebella of Miz1DPOZNes mice, characteristic features of defective autophagy. Our data suggest that Miz1 may link cell growth and ribosome biogenesis to the transcriptional regulation of vesicular transport and autophagy. 1 Theodor Boveri Institute, Biocenter, University of Wu¨rzburg, Am Hubland, 97074 Wu¨rzburg, Germany. 2 Department of Tumor Cell Biology, MS#350, Danny Thomas Research Center, 5006C, St. Jude Children’s Research Hospital, Memphis, Tennessee 38105, USA. 3 Institute for Anatomy and Cell Biology, University of Wu¨rzburg, Koellikerstrasse 6, 97070 Wu¨rzburg, Germany. 4 Comprehensive Cancer Center Mainfranken, Josef-Schneider-Strasse 6, 97080 Wu¨rzburg, Germany. -

Evaluation of a New Method for Large-Scale and Gene-Targeted Next Generation DNA Sequencing in Nonmodel Species
University of Montana ScholarWorks at University of Montana Graduate Student Theses, Dissertations, & Professional Papers Graduate School 2013 Evaluation of a New Method for Large-Scale and Gene-targeted Next Generation DNA Sequencing in Nonmodel Species Ted Cosart The University of Montana Follow this and additional works at: https://scholarworks.umt.edu/etd Let us know how access to this document benefits ou.y Recommended Citation Cosart, Ted, "Evaluation of a New Method for Large-Scale and Gene-targeted Next Generation DNA Sequencing in Nonmodel Species" (2013). Graduate Student Theses, Dissertations, & Professional Papers. 4133. https://scholarworks.umt.edu/etd/4133 This Dissertation is brought to you for free and open access by the Graduate School at ScholarWorks at University of Montana. It has been accepted for inclusion in Graduate Student Theses, Dissertations, & Professional Papers by an authorized administrator of ScholarWorks at University of Montana. For more information, please contact [email protected]. EVALUTATION OF A NEW METHOD FOR LARGE-SCALE AND GENE- TARGETED NEXT GENERATION DNA SEQUENCING IN NONMODEL SPECIES By Ted Cosart BA, University of Montana, Missoula, Montana, 1983 MS, University of Montana, Missoula, Montana, 2006 Dissertation presented in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Individualized, Interdisciplinary Graduate Program The University of Montana Missoula, Montana August, 2013 Approved by: Sandy Ross, Associate Dean of The Graduate School Graduate School Dr. Jesse Johnson, Co-Chair Computer Science Dr. Gordon Luikart, Co-Chair Flathead Biological Station Dr. Jeffrey Good Division of Biological Sciences Dr. William Holben Division of Biological Sciences Dr. Stephen Porcella Rocky Mountain Laboratories, National Institute of Allergy and Infectious Diseases Dr.