Phylogeny of Fomitopsis Pinicola: a Species Complex
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
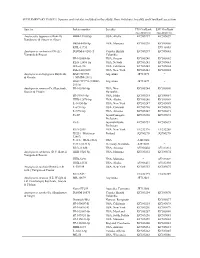
Suppl Table I
SUPPLEMENTARY TABLE I. Species and isolates included in the study, their vouchers, locality and GenBank accession Species Isolate number Locality ITS GenBank LSU GenBank accession no. accession no. Amylocystis lapponica (Romell) HHB-13400-Sp USA, Alaska KC585237 KC585059 Bondartsev & Singer ex Singer OKM-4418-Sp USA, Montana KC585238 KC585060 KHL-11755 − EU118603 Amyloporia carbonica (Overh.) DAOM-F-8281-T Canada, British KC585239 KC585061 Vampola & Pouzar Columbia FP-105585-Sp USA, Oregon KC585240 KC585062 RLG-12496-Sp USA, Nevada KC585241 KC585063 Wilcox-96 USA, California KC585242 KC585064 Zabel-40-GLN USA, New York KC585243 KC585065 Amyloporia nothofaginea Rajchenb. BAFC519794 Argentina JF713078 − & Gorjón (=MMBP-2011) BAFC519796 (=MBP- Argentina JF713079 − 2011a) Amyloporia sinuosa (Fr.) Rajchenb., FP-105386-Sp USA, New KC585244 KC585066 Gorjón & Pildain Hampshire FP-94464-Sp USA, Idaho KC585245 KC585067 HHB-12878-Sp USA, Alaska KC585246 KC585068 L-14130-Sp USA, New York KC585247 KC585069 L-6192-Sp USA, Colorado KC585248 KC585070 L-9792-Sp USA, Arizona KC585249 KC585071 Pa-3C JapanYamagata KC585250 KC585072 Prefecture Pa-3e JapanShizuoka KC585251 KC585073 Prefecture RLG-2538 USA, New York EU232196 EU232288 X725 (=Miettinen- Finland JQ700270 JQ700270 12407) P-115 (=RLG-2538) USA AJ416068 − P-211 (G-214) Germany, Karlsrube AJ345011 − RLG-1182R USA, Arizona AY966450 AY333831 Amyloporia sitchensis (D.V. Baxter) HHB-5320-Sp USA, Montana KC585252 KC585074 Vampola & Pouzar HHB-5298 USA, Montana − AY333829 HHB-12513 USA, Alaska AY966451 AY333830 -

Why Mushrooms Have Evolved to Be So Promiscuous: Insights from Evolutionary and Ecological Patterns
fungal biology reviews 29 (2015) 167e178 journal homepage: www.elsevier.com/locate/fbr Review Why mushrooms have evolved to be so promiscuous: Insights from evolutionary and ecological patterns Timothy Y. JAMES* Department of Ecology and Evolutionary Biology, University of Michigan, Ann Arbor, MI 48109, USA article info abstract Article history: Agaricomycetes, the mushrooms, are considered to have a promiscuous mating system, Received 27 May 2015 because most populations have a large number of mating types. This diversity of mating Received in revised form types ensures a high outcrossing efficiency, the probability of encountering a compatible 17 October 2015 mate when mating at random, because nearly every homokaryotic genotype is compatible Accepted 23 October 2015 with every other. Here I summarize the data from mating type surveys and genetic analysis of mating type loci and ask what evolutionary and ecological factors have promoted pro- Keywords: miscuity. Outcrossing efficiency is equally high in both bipolar and tetrapolar species Genomic conflict with a median value of 0.967 in Agaricomycetes. The sessile nature of the homokaryotic Homeodomain mycelium coupled with frequent long distance dispersal could account for selection favor- Outbreeding potential ing a high outcrossing efficiency as opportunities for choosing mates may be minimal. Pheromone receptor Consistent with a role of mating type in mediating cytoplasmic-nuclear genomic conflict, Agaricomycetes have evolved away from a haploid yeast phase towards hyphal fusions that display reciprocal nuclear migration after mating rather than cytoplasmic fusion. Importantly, the evolution of this mating behavior is precisely timed with the onset of diversification of mating type alleles at the pheromone/receptor mating type loci that are known to control reciprocal nuclear migration during mating. -

Duke Pharmaceutieal Uhiversic}L Nojio 2-522-I, Ktyose, 7Bkyo, 204-8588 Jmpan; Uitiversi(B Box 90338 Durham, IVC27708-O038, CLSA
The JapaneseSocietyJapanese Society for Plant Systematics ISSN 1346-756S Acta Phytotax. Geobot. 54 (2): 99-104 (2003) Bivoriahengduanensis (Lichenized Ascomycota, Parmeliaceae), a New Species from Southern China LI-SONG WANGi, HIROSHI HARADA2, TAKAO NARUI3, CHICITA F. CULBERSON` and WILLIAM L. CULBERSON` iKiinmingfnstitute ofBotarLM ChineseAcadenry qf'Seienee, Ileilongtan, Kiinming funnan 650204, enina; 2 3 History Aoba-eho 955-2, Chuo-ku, Chiba, 260-8682 .lopan; Mbiji Ndtural imseum and institute,Chiba, 4Duke Pharmaceutieal Uhiversic}l Nojio 2-522-I, Ktyose, 7bkyo, 204-8588 Jmpan; Uitiversi(B Box 90338 Durham, IVC27708-O038, CLSA, BtJ)oria hengduanensis L. S. Wang & H. Harada sp. nov., from YUnnan and Sichuan in southern China, is characterized by a pendant thallus, pseudocyphellae spiraling around the branches, meduilary hyphae, and usnic and fumarprotocetraric acids. The closely related B. pseudocupillaris Brodo & D, Hawksw. and B. spiraltfeva Brodo & D. Hawksw, have white pseudocyphellae, andB. tortuosa Brodo & D. L, Hawksw, is sorediate, The new species also differs from all three ofthese North American species by chemistry. Key werds: Alectoria, BT;yoria, chemistry, China, lichenized Ascomycota, new species, OTqpogon, Sichuan, Sulcaria, taxonomy, Minnan Alectorioid lichens are characterized by a fruticose Materials and Methods thallus, terete branches, a distinct cortical layer, an arachnoid medulla without a central axis, a tre- The description ofthe external morphology is based bouxioid photobiont, and lecanorine apothecia. on air-dried material observed under a dissecting They are widely distributed in the world and are rep- microscope, For anatomical description, sections resented in East Asia by Alectoria Ach. in Luyken, rnade with a razor blade were mounted in lac- Br:voria Brodo & D. -

Short-Read Sequencing for Genomic Analysis of the Brown Rot Fungus Fibroporia Radiculosa
Short-Read Sequencing for Genomic Analysis of the Brown Rot Fungus Fibroporia radiculosa Juliet D. Tang,a Andy D. Perkins,b Tad S. Sonstegard,c Steven G. Schroeder,c Shane C. Burgess,d and Susan V. Diehla Forest Products, Mississippi State University, Mississippi State, Mississippi, USAa; Computer Science and Engineering, Mississippi State University, Mississippi State, Mississippi, USAb; USDA ARS Bovine Functional Genomics Laboratory, Beltsville, Maryland, USAc; and Institute for Genomics, Biocomputing, and Biotechnology, Mississippi State University, Mississippi State, Mississippi, USAd The feasibility of short-read sequencing for genomic analysis was demonstrated for Fibroporia radiculosa, a copper-tolerant fungus that causes brown rot decay of wood. The effect of read quality on genomic assembly was assessed by filtering Illumina GAIIx reads from a single run of a paired-end library (75-nucleotide read length and 300-bp fragment size) at three different stringency levels and then assembling each data set with Velvet. A simple approach was devised to determine which filter strin- gency was “best.” Venn diagrams identified the regions containing reads that were used in an assembly but were of a low-enough quality to be removed by a filter. By plotting base quality histograms of reads in this region, we judged whether a filter was too stringent or not stringent enough. Our best assembly had a genome size of 33.6 Mb, an N50 of 65.8 kb for a k-mer of 51, and a maximum contig length of 347 kb. Using GeneMark, 9,262 genes were predicted. TargetP and SignalP analyses showed that among the 1,213 genes with secreted products, 986 had motifs for signal peptides and 227 had motifs for signal anchors. -

Basidiomycota) in Finland
Mycosphere 7 (3): 333–357(2016) www.mycosphere.org ISSN 2077 7019 Article Doi 10.5943/mycosphere/7/3/7 Copyright © Guizhou Academy of Agricultural Sciences Extensions of known geographic distribution of aphyllophoroid fungi (Basidiomycota) in Finland Kunttu P1, Kulju M2, Kekki T3, Pennanen J4, Savola K5, Helo T6 and Kotiranta H7 1University of Eastern Finland, School of Forest Sciences, P.O. Box 111, FI-80101 Joensuu, Finland 2Biodiversity Unit P.O. Box 3000, FI-90014 University of Oulu, Finland 3Jyväskylä University Museum, Natural History Section, P.O. BOX 35, FI-40014 University of Jyväskylä, Finland 4Pentbyntie 1 A 2, FI-10300 Karjaa, Finland 5The Finnish Association for Nature Conservation, Itälahdenkatu 22 b A, FI-00210 Helsinki, Finland 6Erätie 13 C 19, FI-87200 Kajaani, Finland 7Finnish Environment Institute, P.O. Box 140, FI-00251 Helsinki, Finland Kunttu P, Kulju M, Kekki T, Pennanen J, Savola K, Helo T, Kotiranta H 2016 – Extensions of known geographic distribution of aphyllophoroid fungi (Basidiomycota) in Finland. Mycosphere 7(3), 333–357, Doi 10.5943/mycosphere/7/3/7 Abstract This article contributes the knowledge of Finnish aphyllophoroid funga with nationally or regionally new species, and records of rare species. Ceriporia bresadolae, Clavaria tenuipes and Renatobasidium notabile are presented as new aphyllophoroid species to Finland. Ceriporia bresadolae and R. notabile are globally rare species. The records of Ceriporia aurantiocarnescens, Crustomyces subabruptus, Sistotrema autumnale, Trechispora elongata, and Trechispora silvae- ryae are the second in Finland. New records (or localities) are provided for 33 species with no more than 10 records in Finland. In addition, 76 records of aphyllophoroid species are reported as new to some subzones of the boreal vegetation zone in Finland. -

A Phylogenetic Overview of the Antrodia Clade (Basidiomycota, Polyporales)
Mycologia, 105(6), 2013, pp. 1391–1411. DOI: 10.3852/13-051 # 2013 by The Mycological Society of America, Lawrence, KS 66044-8897 A phylogenetic overview of the antrodia clade (Basidiomycota, Polyporales) Beatriz Ortiz-Santana1 phylogenetic studies also have recognized the genera Daniel L. Lindner Amylocystis, Dacryobolus, Melanoporia, Pycnoporellus, US Forest Service, Northern Research Station, Center for Sarcoporia and Wolfiporia as part of the antrodia clade Forest Mycology Research, One Gifford Pinchot Drive, (SY Kim and Jung 2000, 2001; Binder and Hibbett Madison, Wisconsin 53726 2002; Hibbett and Binder 2002; SY Kim et al. 2003; Otto Miettinen Binder et al. 2005), while the genera Antrodia, Botanical Museum, University of Helsinki, PO Box 7, Daedalea, Fomitopsis, Laetiporus and Sparassis have 00014, Helsinki, Finland received attention in regard to species delimitation (SY Kim et al. 2001, 2003; KM Kim et al. 2005, 2007; Alfredo Justo Desjardin et al. 2004; Wang et al. 2004; Wu et al. 2004; David S. Hibbett Dai et al. 2006; Blanco-Dios et al. 2006; Chiu 2007; Clark University, Biology Department, 950 Main Street, Worcester, Massachusetts 01610 Lindner and Banik 2008; Yu et al. 2010; Banik et al. 2010, 2012; Garcia-Sandoval et al. 2011; Lindner et al. 2011; Rajchenberg et al. 2011; Zhou and Wei 2012; Abstract: Phylogenetic relationships among mem- Bernicchia et al. 2012; Spirin et al. 2012, 2013). These bers of the antrodia clade were investigated with studies also established that some of the genera are molecular data from two nuclear ribosomal DNA not monophyletic and several modifications have regions, LSU and ITS. A total of 123 species been proposed: the segregation of Antrodia s.l. -

H. Thorsten Lumbsch VP, Science & Education the Field Museum 1400
H. Thorsten Lumbsch VP, Science & Education The Field Museum 1400 S. Lake Shore Drive Chicago, Illinois 60605 USA Tel: 1-312-665-7881 E-mail: [email protected] Research interests Evolution and Systematics of Fungi Biogeography and Diversification Rates of Fungi Species delimitation Diversity of lichen-forming fungi Professional Experience Since 2017 Vice President, Science & Education, The Field Museum, Chicago. USA 2014-2017 Director, Integrative Research Center, Science & Education, The Field Museum, Chicago, USA. Since 2014 Curator, Integrative Research Center, Science & Education, The Field Museum, Chicago, USA. 2013-2014 Associate Director, Integrative Research Center, Science & Education, The Field Museum, Chicago, USA. 2009-2013 Chair, Dept. of Botany, The Field Museum, Chicago, USA. Since 2011 MacArthur Associate Curator, Dept. of Botany, The Field Museum, Chicago, USA. 2006-2014 Associate Curator, Dept. of Botany, The Field Museum, Chicago, USA. 2005-2009 Head of Cryptogams, Dept. of Botany, The Field Museum, Chicago, USA. Since 2004 Member, Committee on Evolutionary Biology, University of Chicago. Courses: BIOS 430 Evolution (UIC), BIOS 23410 Complex Interactions: Coevolution, Parasites, Mutualists, and Cheaters (U of C) Reading group: Phylogenetic methods. 2003-2006 Assistant Curator, Dept. of Botany, The Field Museum, Chicago, USA. 1998-2003 Privatdozent (Assistant Professor), Botanical Institute, University – GHS - Essen. Lectures: General Botany, Evolution of lower plants, Photosynthesis, Courses: Cryptogams, Biology -

Taxonomy of Bryoria Section Implexae (Parmeliaceae, Lecanoromycetes) in North America and Europe, Based on Chemical, Morphological and Molecular Data
Ann. Bot. Fennici 51: 345–371 ISSN 0003-3847 (print) ISSN 1797-2442 (online) Helsinki 22 September 2014 © Finnish Zoological and Botanical Publishing Board 2014 Taxonomy of Bryoria section Implexae (Parmeliaceae, Lecanoromycetes) in North America and Europe, based on chemical, morphological and molecular data Saara Velmala1,*, Leena Myllys1, Trevor Goward2, Håkon Holien3 & Pekka Halonen4 1) Botanical Museum, Finnish Museum of Natural History, P.O. Box 7, FI-00014 University of Helsinki, Finland (*corresponding author’s e-mail: [email protected]) 2) UBC Herbarium, Beaty Museum, University of British Columbia, Vancouver, BC V6T 1Z4, Canada (mailing address: Enlichened Consulting Ltd., 5369 Clearwater Valley Road, Upper Clearwater, BC V0E 1N1, Canada) 3) Nord-Trøndelag University College, Serviceboks 2501, N-7729 Steinkjer, Norway 4) Botanical Museum, Department of Biology, P.O. Box 3000, FI-90014 University of Oulu, Finland Received 31 Jan. 2014, final version received 13 June 2014, accepted 18 June 2014 Velmala, S., Myllys, L., Goward, T., Holien, H. & Halonen, P. 2014: Taxonomy of Bryoria section Implexae (Parmeliaceae, Lecanoromycetes) in North America and Europe, based on chemical, morphological and molecular data. — Ann. Bot. Fennici 51: 345–371. Ninety-seven ingroup specimens of Bryoria section Implexae (Parmeliaceae, Leca- noromycetes) were studied using molecular, chemical, morphological and geographic characters. The molecular data included nuclear ribosomal markers (ITS, IGS) and the partial glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene. In addition to par- simony analyses, a haplotype network was constructed. Phylogenetic analyses strongly supported the monophyly of the section Implexae. The specimens were grouped into two monophyletic clades. Clade 1 encompassed all esorediate material from North America, whereas Clade 2 included both sorediate North American material and all European material. -

ABSTRACT BOOK Listed Alphabetically by Last Name Of
ABSTRACT BOOK Listed alphabetically by last name of presenting author AOS 2019 Meeting 24-28 June 2019 ORAL PRESENTATIONS Variability in the Use of Acoustic Space Between propensity, renesting intervals, and renest reproductive Two Tropical Forest Bird Communities success of Piping Plovers (Charadrius melodus) by fol- lowing 1,922 nests and 1,785 unique breeding adults Patrick J Hart, Kristina L Paxton, Grace Tredinnick from 2014 2016 in North and South Dakota, USA. The apparent renesting rate was 20%. Renesting propen- When acoustic signals sent from individuals overlap sity declined if reproductive attempts failed during the in frequency or time, acoustic interference and signal brood-rearing stage, nests were depredated, reproduc- masking occurs, which may reduce the receiver’s abil- tive failure occurred later in the breeding season, or ity to discriminate information from the signal. Under individuals had previously renested that year. Addi- the acoustic niche hypothesis (ANH), acoustic space is tionally, plovers were less likely to renest on reservoirs a resource that organisms may compete for, and sig- compared to other habitats. Renesting intervals de- naling behavior has evolved to minimize overlap with clined when individuals had not already renested, were heterospecific calling individuals. Because tropical after second-year adults without prior breeding experi- wet forests have such high bird species diversity and ence, and moved short distances between nest attempts. abundance, and thus high potential for competition for Renesting intervals also decreased if the attempt failed acoustic niche space, they are good places to examine later in the season. Lastly, overall reproductive success the way acoustic space is partitioned. -

Piptoporus Betulinus, Fomitopsis Betulina)
Birch Fungi – Razor Strop, Birch Bracket (Piptoporus betulinus, Fomitopsis betulina) Features - This distinctive fungus only grows on birches, looks like nothing else that grows on birches, and is very common. It is not an aggressive tree killer, but is instead primarily in the business of decomposing dead trees. Birch polypore is present throughout the range of the birches, which grow around the globe in the northern hemisphere. The white-to-brownish fruiting bodies are annual, emerging from the bark of birches in spring and summer, but they deteriorate slowly and are still visible through the winter, though by then they have blackened and are not so attractive. Global Uses – Otzi the Iceman, who lived 5300 years ago, carried two fragments of a fruiting body of Fomitopsis betulina (formerly Piptoporus betulinus). Some scientists believe that Ӧtzi might have used the fungus for medical purposes and, although the idea arouses some controversy, the long tradition of the use of F. betulina in folk medicine is a fact. Infusion from F. betulina fruiting bodies was popular, especially in Russia, Baltic countries, Hungary, Romania for its nutritional and calming properties. Fungal tea was used against various cancer types, as an immunoenhancing, anti-parasitic agent, and a remedy for gastrointestinal disorders. Antiseptic and anti-bleeding dressings made from fresh F. betulina fruiting body were applied to wounds and the powder obtained from dried ones was used as a painkiller. Was used as a razor strop to sharpen fine edged blades. Medicinal Potential – Chemical Constituents: A and C, 1,3-beta-D-glucopyranan, B ergosta-7,22-dien-3- ol, fungisterol, ergosterol, agaric acid, dehydrotumulosic acid, ungalinic acid, betulinic acid, and tumulosic acid. -

Identification, Effects and Management of 5 Types of Decay Organisms Found in Seattle Parks
Identification, Effects and Management of 5 types of decay organisms found in Seattle Parks Chris Rippey, Arborist [email protected] • Third generation Arborist • Grew up in the bay area of California. • Was 16 when I started working with my dad in tree care • I fell in love with tree work, not trees • Managed the preventative tree maintenance programs at Stanford University for 14 years. • Moved to Washington and began working for Seattle Parks 2 ½ years ago Seattle Parks System - 6,412 Total Acres - 4,016 Acres of Developed Park - 2,396 Acres of Natural Area - 480 Parks - >300,000 trees - >16,000 trees in our tree inventory Seward Park 1920 Ravenna Park 1922 What are we focusing on? - 171,615 trees in our Buffer Zone. - Buffer Zone is a 50’ buffer around high use areas like beaches, paved roads and trails, playgrounds…etc) - Buffer Zones are on average 56% of a given park Tree Risk Inspections Terms & Matrix TERM DEFINITION Likelihood of failure and impacts Imminent Failure has started or is most likely to occur in the near future even if there is no weather forces/rare occurrence. Will fail in a storm. Probable Failure may be expected under normal weather within a time frame. Likely to fail in a severe storm. Possible Failure could occur, but is unlikely during normal weather. May fail in a severe storm. Improbable Tree or branch failure not likely under normal conditions and may not fail in severe weather within a time frame. Risk rating High Failed tree or part will likely impact a target. -

The Cardioprotective Properties of Agaricomycetes Mushrooms Growing in the Territory of Armenia (Review) Susanna Badalyan, Anush Barkhudaryan, Sylvie Rapior
The Cardioprotective Properties of Agaricomycetes Mushrooms Growing in the Territory of Armenia (Review) Susanna Badalyan, Anush Barkhudaryan, Sylvie Rapior To cite this version: Susanna Badalyan, Anush Barkhudaryan, Sylvie Rapior. The Cardioprotective Properties of Agari- comycetes Mushrooms Growing in the Territory of Armenia (Review). International Journal of Medic- inal Mushrooms, Begell House, 2021, 23 (5), pp.21-31. 10.1615/IntJMedMushrooms.2021038280. hal-03202984 HAL Id: hal-03202984 https://hal.umontpellier.fr/hal-03202984 Submitted on 20 Apr 2021 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. The Cardioprotective Properties of Agaricomycetes Mushrooms Growing in the territory of Armenia (Review) Susanna M. Badalyan 1, Anush Barkhudaryan 2, Sylvie Rapior 3 1Laboratory of Fungal Biology and Biotechnology, Institute of Pharmacy, Department of Biomedicine, Yerevan State University, Yerevan, Armenia; 2Department of Cardiology, Clinic of General and Invasive Cardiology, University Hospital № 1, Yerevan State Medical University, Yerevan, Armenia;