Microarray Comparative Genomic Hybridisation Analysis of Intraocular Uveal Melanomas Identifies Distinctive Imbalances Associated with Loss of Chromosome 3
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Deregulated Gene Expression Pathways in Myelodysplastic Syndrome Hematopoietic Stem Cells
Leukemia (2010) 24, 756–764 & 2010 Macmillan Publishers Limited All rights reserved 0887-6924/10 $32.00 www.nature.com/leu ORIGINAL ARTICLE Deregulated gene expression pathways in myelodysplastic syndrome hematopoietic stem cells A Pellagatti1, M Cazzola2, A Giagounidis3, J Perry1, L Malcovati2, MG Della Porta2,MJa¨dersten4, S Killick5, A Verma6, CJ Norbury7, E Hellstro¨m-Lindberg4, JS Wainscoat1 and J Boultwood1 1LRF Molecular Haematology Unit, NDCLS, John Radcliffe Hospital, Oxford, UK; 2Department of Hematology Oncology, University of Pavia Medical School, Fondazione IRCCS Policlinico San Matteo, Pavia, Italy; 3Medizinische Klinik II, St Johannes Hospital, Duisburg, Germany; 4Division of Hematology, Department of Medicine, Karolinska Institutet, Stockholm, Sweden; 5Department of Haematology, Royal Bournemouth Hospital, Bournemouth, UK; 6Albert Einstein College of Medicine, Bronx, NY, USA and 7Sir William Dunn School of Pathology, University of Oxford, Oxford, UK To gain insight into the molecular pathogenesis of the the World Health Organization.6,7 Patients with refractory myelodysplastic syndromes (MDS), we performed global gene anemia (RA) with or without ringed sideroblasts, according to expression profiling and pathway analysis on the hemato- poietic stem cells (HSC) of 183 MDS patients as compared with the the French–American–British classification, were subdivided HSC of 17 healthy controls. The most significantly deregulated based on the presence or absence of multilineage dysplasia. In pathways in MDS include interferon signaling, thrombopoietin addition, patients with RA with excess blasts (RAEB) were signaling and the Wnt pathways. Among the most signifi- subdivided into two categories, RAEB1 and RAEB2, based on the cantly deregulated gene pathways in early MDS are immuno- percentage of bone marrow blasts. -

Intratumor Heterogeneity: Novel Approaches for Resolving Genomic Architecture and Clonal Evolution Ravi G
Published OnlineFirst June 8, 2017; DOI: 10.1158/1541-7786.MCR-17-0070 Review Molecular Cancer Research Intratumor Heterogeneity: Novel Approaches for Resolving Genomic Architecture and Clonal Evolution Ravi G. Gupta and Robert A. Somer Abstract High-throughput genomic technologies have revealed a full mutational landscape of tumors could help reconstruct remarkably complex portrait of intratumor heterogeneity in their phylogenetic trees and trace the subclonal origins of cancer and have shown that tumors evolve through a reiterative therapeutic resistance, relapsed disease, and distant metastases, process of genetic diversification and clonal selection. This the major causes of cancer-related mortality. Real-time assess- discovery has challenged the classical paradigm of clonal ment of the tumor subclonal architecture, however, remains dominance and brought attention to subclonal tumor cell limited by the high rate of errors produced by most genome- populations that contribute to the cancer phenotype. Dynamic wide sequencing methods as well as the practical difficulties evolutionary models may explain how these populations grow associated with serial tumor genotyping in patients. This review within the ecosystem of tissues, including linear, branching, focuses on novel approaches to mitigate these challenges using neutral, and punctuated patterns. Recent evidence in breast bulk tumor, liquid biopsies, single-cell analysis, and deep cancer favors branching and punctuated evolution driven by sequencing techniques. The origins of intratumor heterogene- genome instability as well as nongenetic sources of heteroge- ity and the clinical, diagnostic, and therapeutic consequences neity, such as epigenetic variation, hierarchal tumor cell orga- in breast cancer are also explored. Mol Cancer Res; 15(9); 1127–37. nization, and subclonal cell–cell interactions. -

PLXNB1 (Plexin
Atlas of Genetics and Cytogenetics in Oncology and Haematology OPEN ACCESS JOURNAL AT INIST-CNRS Gene Section Mini Review PLXNB1 (plexin B1) José Javier Gómez-Román, Montserrat Nicolas Martínez, Servando Lazuén Fernández, José Fernando Val-Bernal Department of Anatomical Pathology, Marques de Valdecilla University Hospital, Medical Faculty, University of Cantabria, Santander, Spain (JJGR, MN, SL, JFVB) Published in Atlas Database: March 2009 Online updated version: http://AtlasGeneticsOncology.org/Genes/PLXNB1ID43413ch3p21.html DOI: 10.4267/2042/44702 This work is licensed under a Creative Commons Attribution-Noncommercial-No Derivative Works 2.0 France Licence. © 2010 Atlas of Genetics and Cytogenetics in Oncology and Haematology Identity Pseudogene No. Other names: KIAA0407; MGC149167; OTTHUMP00000164806; PLEXIN-B1; PLXN5; SEP Protein HGNC (Hugo): PLXNB1 Location: 3p21.31 Description Local order: The Plexin B1 gene is located between 2135 Amino acids (AA). Plexins are receptors for axon FBXW12 and CCDC51 genes. molecular guidance molecules semaphorins. Plexin signalling is important in pathfinding and patterning of both neurons and developing blood vessels. Plexin-B1 is a surface cell receptor. When it binds to its ligand SEMA4D it activates several pathways by binding of cytoplasmic ligands, like RHOA activation and subsequent changes of the actin cytoskeleton, axon guidance, invasive growth and cell migration. It monomers and heterodimers with PLXNB2 after proteolytic processing. Binds RAC1 that has been activated by GTP binding. It binds PLXNA1 and by similarity ARHGEF11, Note ARHGEF12, ERBB2, MET, MST1R, RND1, NRP1 Size: 26,200 bases. and NRP2. Orientation: minus strand. This family features the C-terminal regions of various plexins. The cytoplasmic region, which has been called DNA/RNA a SEX domain in some members of this family is involved in downstream signalling pathways, by Description interaction with proteins such as Rac1, RhoD, Rnd1 and other plexins. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Noninvasive Sleep Monitoring in Large-Scale Screening of Knock-Out Mice
bioRxiv preprint doi: https://doi.org/10.1101/517680; this version posted January 11, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. Noninvasive sleep monitoring in large-scale screening of knock-out mice reveals novel sleep-related genes Shreyas S. Joshi1*, Mansi Sethi1*, Martin Striz1, Neil Cole2, James M. Denegre2, Jennifer Ryan2, Michael E. Lhamon3, Anuj Agarwal3, Steve Murray2, Robert E. Braun2, David W. Fardo4, Vivek Kumar2, Kevin D. Donohue3,5, Sridhar Sunderam6, Elissa J. Chesler2, Karen L. Svenson2, Bruce F. O'Hara1,3 1Dept. of Biology, University of Kentucky, Lexington, KY 40506, USA, 2The Jackson Laboratory, Bar Harbor, ME 04609, USA, 3Signal solutions, LLC, Lexington, KY 40503, USA, 4Dept. of Biostatistics, University of Kentucky, Lexington, KY 40536, USA, 5Dept. of Electrical and Computer Engineering, University of Kentucky, Lexington, KY 40506, USA. 6Dept. of Biomedical Engineering, University of Kentucky, Lexington, KY 40506, USA. *These authors contributed equally Address for correspondence and proofs: Shreyas S. Joshi, Ph.D. Dept. of Biology University of Kentucky 675 Rose Street 101 Morgan Building Lexington, KY 40506 U.S.A. Phone: (859) 257-2805 FAX: (859) 257-1717 Email: [email protected] Running title: Sleep changes in knockout mice bioRxiv preprint doi: https://doi.org/10.1101/517680; this version posted January 11, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. -

Molecular Associations and Clinical Significance of Raps In
ORIGINAL RESEARCH published: 21 June 2021 doi: 10.3389/fmolb.2021.677979 Molecular Associations and Clinical Significance of RAPs in Hepatocellular Carcinoma Sarita Kumari 1†, Mohit Arora 2†, Jay Singh 1, Lokesh K. Kadian 2, Rajni Yadav 3, Shyam S. Chauhan 2* and Anita Chopra 1* 1Laboratory Oncology Unit, Dr. BRA-IRCH, All India Institute of Medical Sciences, New Delhi, India, 2Department of Biochemistry, All India Institute of Medical Sciences, New Delhi, India, 3Department of Pathology, All India Institute of Medical Sciences, New Delhi, India Hepatocellular carcinoma (HCC) is an aggressive gastrointestinal malignancy with a high rate of mortality. Multiple studies have individually recognized members of RAP gene family as critical regulators of tumor progression in several cancers, including hepatocellular carcinoma. These studies suffer numerous limitations including a small sample size and lack of analysis of various clinicopathological and molecular Edited by: Veronica Aran, features. In the current study, we utilized authoritative multi-omics databases to Instituto Estadual do Cérebro Paulo determine the association of RAP gene family expression and detailed molecular and Niemeyer (IECPN), Brazil clinicopathological features in hepatocellular carcinoma (HCC). All five RAP genes Reviewed by: were observed to harbor dysregulated expression in HCC compared to normal liver Pooja Panwalkar, Weill Cornell Medicine, United States tissues. RAP2A exhibited strongest ability to differentiate tumors from the normal Jasminka Omerovic, tissues. RAP2A expression was associated with progressive tumor grade, TP53 and University of Split, Croatia CTNNB1 mutation status. Additionally, RAP2A expression was associated with the *Correspondence: Anita Chopra alteration of its copy numbers and DNA methylation. RAP2A also emerged as an [email protected] independent marker for patient prognosis. -
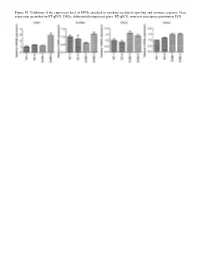
Figure S1. Validation of the Expression Level of Degs Enriched in Cytokine‑Mediated Signaling and Immune Response
Figure S1. Validation of the expression level of DEGs enriched in cytokine‑mediated signaling and immune response. Gene expression quantified by RT‑qPCR. DEGs, differentially expressed genes; RT‑qPCR, reverse‑transcription quantitative PCR. Figure S2. Validation of STAU1‑regulated AS events. IGV‑Sashimi plot revealed (A‑C) three A5SS AS events in three different genes. Reads distribution of each AS event was plotted in the left panel with the transcripts of each gene shown below. The sche‑ matic diagrams depict the structures of two AS events, AS1 (purple line) and AS2 (green line). The exon sequences are denoted by black boxes, the intron sequences by a horizontal line (right panel). RNA‑seq quantification and RT‑qPCR validation of ASEs are presented in the panels on the right. STAU1, double‑stranded RNA‑binding protein Staufen homolog 1; AS, alternative splicing; A5SS, alternative 5'splice site; RNA‑seq, RNA sequencing; RT‑qPCR, reverse‑transcription quantitative PCR. Error bars represent mean ± SEM. *P<0.05. Table SI. Primers used in gene validation experiments. IFIT2‑F CAGCCTACGGCAACTAAA IFIT2‑R GAGCCTTCTCAAAGCACA IFIT3‑F ACACCAAACAATGGCTAC IFIT3‑R TGGACAAACCCTCTAAAC OASL‑F AATGGTGACCGTGATGGG OASL‑R ACCTGAGGATGGAGCAGAG IFI27‑F TTCACTGCGGCGGGAATC IFI27‑R TGGCTGCTATGGAGGACGAG S1PR4‑F TGCTGAAGACGGTGCTGATG S1PR4‑R TGCGGAAGGAGTAGATGATGG CCL5‑F ACGACTGCTGGGTTGGAG CCL5‑R ACCCTGCTGCTTTGCCTA CCL2‑F CTAACCCAGAAACATCCAAT CCL2‑R GCTATGAGCAGCAGGCAC CD44‑F TGGAGGACAGAAAGCCAAGT CD44‑R TTCGCAATGAAACAATCAGTAG PLEKHG2‑M/As‑F CCAAAAGTAAGCCTGTCC PLEKHG2‑M‑R -

The Function and Evolution of C2H2 Zinc Finger Proteins and Transposons
The function and evolution of C2H2 zinc finger proteins and transposons by Laura Francesca Campitelli A thesis submitted in conformity with the requirements for the degree of Doctor of Philosophy Department of Molecular Genetics University of Toronto © Copyright by Laura Francesca Campitelli 2020 The function and evolution of C2H2 zinc finger proteins and transposons Laura Francesca Campitelli Doctor of Philosophy Department of Molecular Genetics University of Toronto 2020 Abstract Transcription factors (TFs) confer specificity to transcriptional regulation by binding specific DNA sequences and ultimately affecting the ability of RNA polymerase to transcribe a locus. The C2H2 zinc finger proteins (C2H2 ZFPs) are a TF class with the unique ability to diversify their DNA-binding specificities in a short evolutionary time. C2H2 ZFPs comprise the largest class of TFs in Mammalian genomes, including nearly half of all Human TFs (747/1,639). Positive selection on the DNA-binding specificities of C2H2 ZFPs is explained by an evolutionary arms race with endogenous retroelements (EREs; copy-and-paste transposable elements), where the C2H2 ZFPs containing a KRAB repressor domain (KZFPs; 344/747 Human C2H2 ZFPs) are thought to diversify to bind new EREs and repress deleterious transposition events. However, evidence of the gain and loss of KZFP binding sites on the ERE sequence is sparse due to poor resolution of ERE sequence evolution, despite the recent publication of binding preferences for 242/344 Human KZFPs. The goal of my doctoral work has been to characterize the Human C2H2 ZFPs, with specific interest in their evolutionary history, functional diversity, and coevolution with LINE EREs. -

Role and Regulation of the P53-Homolog P73 in the Transformation of Normal Human Fibroblasts
Role and regulation of the p53-homolog p73 in the transformation of normal human fibroblasts Dissertation zur Erlangung des naturwissenschaftlichen Doktorgrades der Bayerischen Julius-Maximilians-Universität Würzburg vorgelegt von Lars Hofmann aus Aschaffenburg Würzburg 2007 Eingereicht am Mitglieder der Promotionskommission: Vorsitzender: Prof. Dr. Dr. Martin J. Müller Gutachter: Prof. Dr. Michael P. Schön Gutachter : Prof. Dr. Georg Krohne Tag des Promotionskolloquiums: Doktorurkunde ausgehändigt am Erklärung Hiermit erkläre ich, dass ich die vorliegende Arbeit selbständig angefertigt und keine anderen als die angegebenen Hilfsmittel und Quellen verwendet habe. Diese Arbeit wurde weder in gleicher noch in ähnlicher Form in einem anderen Prüfungsverfahren vorgelegt. Ich habe früher, außer den mit dem Zulassungsgesuch urkundlichen Graden, keine weiteren akademischen Grade erworben und zu erwerben gesucht. Würzburg, Lars Hofmann Content SUMMARY ................................................................................................................ IV ZUSAMMENFASSUNG ............................................................................................. V 1. INTRODUCTION ................................................................................................. 1 1.1. Molecular basics of cancer .......................................................................................... 1 1.2. Early research on tumorigenesis ................................................................................. 3 1.3. Developing -

Intra-Tumor Heterogeneity from a Cancer Stem Cell Perspective Pramudita R
Prasetyanti and Medema Molecular Cancer (2017) 16:41 DOI 10.1186/s12943-017-0600-4 REVIEW Open Access Intra-tumor heterogeneity from a cancer stem cell perspective Pramudita R. Prasetyanti1,2 and Jan Paul Medema1,2,3* Abstract Tumor heterogeneity represents an ongoing challenge in the field of cancer therapy. Heterogeneity is evident between cancers from different patients (inter-tumor heterogeneity) and within a single tumor (intra-tumor heterogeneity). The latter includes phenotypic diversity such as cell surface markers, (epi)genetic abnormality, growth rate, apoptosis and other hallmarks of cancer that eventually drive disease progression and treatment failure. Cancer stem cells (CSCs) have been put forward to be one of the determining factors that contribute to intra-tumor heterogeneity. However, recent findings have shown that the stem-like state in a given tumor cell is a plastic quality. A corollary to this view is that stemness traits can be acquired via (epi)genetic modification and/or interaction with the tumor microenvironment (TME). Here we discuss factors contributing to this CSC heterogeneity and the potential implications for cancer therapy. Keywords: Tumor heterogeneity, Cancer stem cell, Stemness, Tumor microenvironment Background tumor and microenvironment heterogeneity determine A tumor is a heterogeneous population of cells, containing the fitness of the tumor and as such, are likely to be transformed cancer cells, supportive cells and tumor- crucial factors in treatment success. infiltrating cells. This intra-tumor heterogeneity is further Two models have been proposed to account for hetero- enhanced by clonal variation and microenvironmental geneity within a tumor. In the clonal evolution model, influences on cancer cells, which also do not represent a stochastic mutations in individual tumor cells serve as homogeneous set of cells. -

Human Induced Pluripotent Stem Cell–Derived Podocytes Mature Into Vascularized Glomeruli Upon Experimental Transplantation
BASIC RESEARCH www.jasn.org Human Induced Pluripotent Stem Cell–Derived Podocytes Mature into Vascularized Glomeruli upon Experimental Transplantation † Sazia Sharmin,* Atsuhiro Taguchi,* Yusuke Kaku,* Yasuhiro Yoshimura,* Tomoko Ohmori,* ‡ † ‡ Tetsushi Sakuma, Masashi Mukoyama, Takashi Yamamoto, Hidetake Kurihara,§ and | Ryuichi Nishinakamura* *Department of Kidney Development, Institute of Molecular Embryology and Genetics, and †Department of Nephrology, Faculty of Life Sciences, Kumamoto University, Kumamoto, Japan; ‡Department of Mathematical and Life Sciences, Graduate School of Science, Hiroshima University, Hiroshima, Japan; §Division of Anatomy, Juntendo University School of Medicine, Tokyo, Japan; and |Japan Science and Technology Agency, CREST, Kumamoto, Japan ABSTRACT Glomerular podocytes express proteins, such as nephrin, that constitute the slit diaphragm, thereby contributing to the filtration process in the kidney. Glomerular development has been analyzed mainly in mice, whereas analysis of human kidney development has been minimal because of limited access to embryonic kidneys. We previously reported the induction of three-dimensional primordial glomeruli from human induced pluripotent stem (iPS) cells. Here, using transcription activator–like effector nuclease-mediated homologous recombination, we generated human iPS cell lines that express green fluorescent protein (GFP) in the NPHS1 locus, which encodes nephrin, and we show that GFP expression facilitated accurate visualization of nephrin-positive podocyte formation in -

Suppl Fig 2 PB2 EUCOMM V3
Maier et al. Supplementary material Semaphorin 4C and 4G are Plexin-B2 ligands required in cerebellar development 2 Supplementary tables 4 Supplementary figures 1 Supplementary Table 1. Exencephaly and postnatal survival of Sema4C mutants (on C57BL/6 background) Genotype Expected frequency Observed frequency Exencephaly Embryonic stages (n=47)* Wild type 25% 25% (12/47) 0% Sema4c+/- 50% 45% (21/47) 0% Sema4c-/- 25% 30% (14/47) 36% (5/14) Postnatal animals (n=417)* Wild type 25% 27% (113/417) Sema4c+/- 50% 61% (253/417) Sema4c-/- 25% 12% (51/417) * Offspring from Sema4c+/- x +/- matings. Embryonic stages scored at E15-E18, postnatal animals scored at P21. 2 Supplementary Table 2. Cerebellar phenotypes of Sema4C and Sema4G mutants (on CD-1 outbred background) Ectopic Fusion of Gap in Normal Gap in IGL granule cells lobules IGL of cerebellum of lobule II in molecular VIII/IX* lobule X Genotype n layer weak strong Wild type 17 100% 0% 0% 0% 0% 0% Sema4c+/- 11 70% 10% 0% 10% 0% 10% Sema4c-/- 10 20% 70% 0% 40% 0% 40% Sema4c+/-; Sema4g+/- 16 55% 18% 0% 18% 0% 18% Sema4c+/-; Sema4g-/- 13 40% 20% 0% 20% 0% 40% Sema4c-/-; Sema4g+/- 10 20% 30% 0% 50% 0% 60% Sema4c-/-; Sema4g-/- 11 0% 55% 0% 55% 55% 82% *Fusions of lobules VIII and IX were scored as “weak” when ectopic granule cells formed a band of cells at the fusion line, and as “strong” when a continuous bridge of granule cells connected the IGL of the two lobules. 3 Supplementary figure legends Maier et al. Suppl. Fig.