Integrative Prediction of Gene Expression with Chromatin Accessibility and Conformation Data Florian Schmidt1,2,3,4†, Fabian Kern1,3,5† and Marcel H
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Mediator Head Subcomplex Med11/22 Contains a Common Helix Bundle
Published online 15 April 2011 Nucleic Acids Research, 2011, Vol. 39, No. 14 6291–6304 doi:10.1093/nar/gkr229 Mediator head subcomplex Med11/22 contains a common helix bundle building block with a specific function in transcription initiation complex stabilization Martin Seizl, Laurent Larivie` re, Toni Pfaffeneder, Larissa Wenzeck and Patrick Cramer* Gene Center and Department of Biochemistry, Center for Integrated Protein Science Munich (CIPSM), Ludwig-Maximilians-Universita¨ t (LMU) Mu¨ nchen, Feodor-Lynen-Strasse 25, 81377 Munich, Germany Received February 24, 2011; Revised and Accepted March 29, 2011 ABSTRACT Mediator undergoes conformational changes, which trig- Mediator is a multiprotein co-activator of RNA poly- ger its interaction with Pol II and promote pre-initiation merase (Pol) II transcription. Mediator contains a complex (PIC) formation (7). Mediator was purified from yeast (8,9), metazoans conserved core that comprises the ‘head’ and (10,11) and plants (12). Comparative genomics identified ‘middle’ modules. We present here a structure– an ancient 17-subunit Mediator core that is conserved function analysis of the essential Med11/22 hetero- in eukaryotes (13,14). Mediator from the yeast dimer, a part of the head module. Med11/22 forms a Saccharomyces cerevisiae (Sc) is a 1.4 MDa multiprotein conserved four-helix bundle domain with C-terminal complex comprising 25 subunits, of which 11 are essential extensions, which bind the central head subunit and 22 are at least partially conserved. Based on electron Med17. A highly conserved patch on the bundle microscopy and biochemical analysis, mediator subunits surface is required for stable transcription pre- were suggested to reside in four flexibly linked modules, initiation complex formation on a Pol II promoter the head, middle, tail and kinase module (15–17). -

Transcriptomic Analysis of Pluripotent Stem Cells: Insights Into Health and Disease Jia-Chi Yeo1,2 and Huck-Hui Ng1,2,3,4,5,*
Yeo and Ng Genome Medicine 2011, 3:68 http://genomemedicine.com/content/3/10/68 REVIEW Transcriptomic analysis of pluripotent stem cells: insights into health and disease Jia-Chi Yeo1,2 and Huck-Hui Ng1,2,3,4,5,* Abstract types, termed ‘pluripotency’, allows researchers to study early mammalian development in an artificial setting and Embryonic stem cells (ESCs) and induced pluripotent offers opportunities for regenerative medicine, whereby stem cells (iPSCs) hold tremendous clinical potential ESCs could generate clinically relevant cell types for because of their ability to self-renew, and to tissue repair. However, this same malleability of ESCs dierentiate into all cell types of the body. This unique also renders it a challenge to obtain in vitro differentiation capacity of ESCs and iPSCs to form all cell lineages of ESCs to specific cell types at high efficacy. erefore, is termed pluripotency. While ESCs and iPSCs are harnessing the full potential of ESCs requires an in-depth pluripotent and remarkably similar in appearance, understanding of the factors and mechanisms regulating whether iPSCs truly resemble ESCs at the molecular ESC pluripotency and cell lineage decisions. level is still being debated. Further research is therefore Early studies on ESCs led to the discovery of the core needed to resolve this issue before iPSCs may be safely pluripotency factors Oct4, Sox2 and Nanog [1], and, applied in humans for cell therapy or regenerative increasingly, the use of genome-level screening assays has medicine. Nevertheless, the use of iPSCs as an in vitro revealed new insights by uncovering additional trans- human genetic disease model has been useful in cription factors, transcriptional cofactors and chromatin studying the molecular pathology of complex genetic remodeling complexes involved in the maintenance of diseases, as well as facilitating genetic or drug screens. -

The Title of the Dissertation
UNIVERSITY OF CALIFORNIA SAN DIEGO Novel network-based integrated analyses of multi-omics data reveal new insights into CD8+ T cell differentiation and mouse embryogenesis A dissertation submitted in partial satisfaction of the requirements for the degree Doctor of Philosophy in Bioinformatics and Systems Biology by Kai Zhang Committee in charge: Professor Wei Wang, Chair Professor Pavel Arkadjevich Pevzner, Co-Chair Professor Vineet Bafna Professor Cornelis Murre Professor Bing Ren 2018 Copyright Kai Zhang, 2018 All rights reserved. The dissertation of Kai Zhang is approved, and it is accept- able in quality and form for publication on microfilm and electronically: Co-Chair Chair University of California San Diego 2018 iii EPIGRAPH The only true wisdom is in knowing you know nothing. —Socrates iv TABLE OF CONTENTS Signature Page ....................................... iii Epigraph ........................................... iv Table of Contents ...................................... v List of Figures ........................................ viii List of Tables ........................................ ix Acknowledgements ..................................... x Vita ............................................. xi Abstract of the Dissertation ................................. xii Chapter 1 General introduction ............................ 1 1.1 The applications of graph theory in bioinformatics ......... 1 1.2 Leveraging graphs to conduct integrated analyses .......... 4 1.3 References .............................. 6 Chapter 2 Systematic -

Regulation of Adult Neurogenesis in Mammalian Brain
International Journal of Molecular Sciences Review Regulation of Adult Neurogenesis in Mammalian Brain 1,2, 3, 3,4 Maria Victoria Niklison-Chirou y, Massimiliano Agostini y, Ivano Amelio and Gerry Melino 3,* 1 Centre for Therapeutic Innovation (CTI-Bath), Department of Pharmacy & Pharmacology, University of Bath, Bath BA2 7AY, UK; [email protected] 2 Blizard Institute of Cell and Molecular Science, Barts and the London School of Medicine and Dentistry, Queen Mary University of London, London E1 2AT, UK 3 Department of Experimental Medicine, TOR, University of Rome “Tor Vergata”, 00133 Rome, Italy; [email protected] (M.A.); [email protected] (I.A.) 4 School of Life Sciences, University of Nottingham, Nottingham NG7 2HU, UK * Correspondence: [email protected] These authors contributed equally to this work. y Received: 18 May 2020; Accepted: 7 July 2020; Published: 9 July 2020 Abstract: Adult neurogenesis is a multistage process by which neurons are generated and integrated into existing neuronal circuits. In the adult brain, neurogenesis is mainly localized in two specialized niches, the subgranular zone (SGZ) of the dentate gyrus and the subventricular zone (SVZ) adjacent to the lateral ventricles. Neurogenesis plays a fundamental role in postnatal brain, where it is required for neuronal plasticity. Moreover, perturbation of adult neurogenesis contributes to several human diseases, including cognitive impairment and neurodegenerative diseases. The interplay between extrinsic and intrinsic factors is fundamental in regulating neurogenesis. Over the past decades, several studies on intrinsic pathways, including transcription factors, have highlighted their fundamental role in regulating every stage of neurogenesis. However, it is likely that transcriptional regulation is part of a more sophisticated regulatory network, which includes epigenetic modifications, non-coding RNAs and metabolic pathways. -

Variation in Protein Coding Genes Identifies Information
bioRxiv preprint doi: https://doi.org/10.1101/679456; this version posted June 21, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Animal complexity and information flow 1 1 2 3 4 5 Variation in protein coding genes identifies information flow as a contributor to 6 animal complexity 7 8 Jack Dean, Daniela Lopes Cardoso and Colin Sharpe* 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 Institute of Biological and Biomedical Sciences 25 School of Biological Science 26 University of Portsmouth, 27 Portsmouth, UK 28 PO16 7YH 29 30 * Author for correspondence 31 [email protected] 32 33 Orcid numbers: 34 DLC: 0000-0003-2683-1745 35 CS: 0000-0002-5022-0840 36 37 38 39 40 41 42 43 44 45 46 47 48 49 Abstract bioRxiv preprint doi: https://doi.org/10.1101/679456; this version posted June 21, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Animal complexity and information flow 2 1 Across the metazoans there is a trend towards greater organismal complexity. How 2 complexity is generated, however, is uncertain. Since C.elegans and humans have 3 approximately the same number of genes, the explanation will depend on how genes are 4 used, rather than their absolute number. -

Effects of Downregulating GLIS1 Transcript On
Journal of Reproduction and Development, Vol. 61, No 5, 2015 —Original Article— Effects of downregulating GLIS1 transcript on preimplantation development and gene expression of bovine embryos Kazuki TAKAHASHI1), Nobuyuki SAKURAI2), Natsuko EMURA1), Tsutomu HASHIZUME1, 2) and Ken SAWAI1, 2) 1)Faculty of Agriculture, Iwate University, Iwate 020-8550, Japan 2)The United Graduate School of Agricultural Sciences, Iwate University, Iwate 020-8550, Japan Abstract. Krüppel-like protein Gli-similar 1 (GLIS1) is known as a direct reprogramming factor for the generation of induced pluripotent stem cells. The objective of this study was to investigate the role of GLIS1 in the preimplantation development of bovine embryos. GLIS1 transcripts in in vitro-matured oocytes and 1-cell to 4-cell stage embryos were detected, but they were either absent or at trace levels at the 8-cell to blastocyst stages. We attempted GLIS1 downregulation of bovine early embryos by RNA interference and evaluated developmental competency and gene transcripts, which are involved in zygotic gene activation (ZGA) in GLIS1-downregulated embryos. Injection of specific siRNA resulted in a distinct decrease inGLIS1 transcript in bovine embryos at the 4-cell stage. Although the bovine embryos injected with GLIS1-siRNA could develop to the 16-cell stage, these embryos had difficulty in developing beyond the 32-cell stage. Gene transcripts of PDHA1 and HSPA8, which are transcribed after ZGA, showed lower level in GLIS1 downregulated embryos. It is possible that GLIS1- downregulated embryos fail to initiate ZGA. Our results indicated that GLIS1 is an important factor for the preimplantation development of bovine embryos. Key words: Bovine embryo, Early development, Gene expression, GLIS1, RNA interference (J. -

The Suppressive Effects of 1,25-Dihydroxyvitamin D3 and Vitamin D Receptor on Brown Adipocyte Differentiation and Mitochondrial Respiration
University of Tennessee, Knoxville TRACE: Tennessee Research and Creative Exchange Masters Theses Graduate School 8-2014 The Suppressive Effects of 1,25-dihydroxyvitamin D3 and Vitamin D Receptor on Brown Adipocyte Differentiation and Mitochondrial Respiration Carolyn Jeanne Ricciardi University of Tennessee - Knoxville, [email protected] Follow this and additional works at: https://trace.tennessee.edu/utk_gradthes Part of the Molecular, Genetic, and Biochemical Nutrition Commons Recommended Citation Ricciardi, Carolyn Jeanne, "The Suppressive Effects of 1,25-dihydroxyvitamin D3 and Vitamin D Receptor on Brown Adipocyte Differentiation and Mitochondrial Respiration. " Master's Thesis, University of Tennessee, 2014. https://trace.tennessee.edu/utk_gradthes/2876 This Thesis is brought to you for free and open access by the Graduate School at TRACE: Tennessee Research and Creative Exchange. It has been accepted for inclusion in Masters Theses by an authorized administrator of TRACE: Tennessee Research and Creative Exchange. For more information, please contact [email protected]. To the Graduate Council: I am submitting herewith a thesis written by Carolyn Jeanne Ricciardi entitled "The Suppressive Effects of 1,25-dihydroxyvitamin D3 and Vitamin D Receptor on Brown Adipocyte Differentiation and Mitochondrial Respiration." I have examined the final electronic copy of this thesis for form and content and recommend that it be accepted in partial fulfillment of the equirr ements for the degree of Master of Science, with a major in Nutrition. -
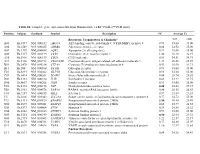
Table S1. Complete Gene Expression Data from Human Diabetes RT² Profiler™ PCR Array Receptors, Transporters & Channels* A
Table S1. Complete gene expression data from Human Diabetes RT² Profiler™ PCR Array Position Unigene GenBank Symbol Description FC Average Ct Receptors, Transporters & Channels* NGT GDM A01 Hs,5447 NM_000352 ABCC8 ATP-binding cassette, sub-family C (CFTR/MRP), member 8 0.93 35.00 35.00 A04 0Hs,2549 NM_000025 ADRB3 Adrenergic, beta-3-, receptor 0.88 34.92 35.00 A07 Hs,1307 NM_000486 AQP2 Aquaporin 2 (collecting duct) 0.93 35.00 35.00 A09 30Hs,5117 NM_001123 CCR2 Chemokine (C-C motif) receptor 2 1.00 26.28 26.17 A10 94Hs,5916 396NM_006139 CD28 CD28 molecule 0.81 34.51 34.71 A11 29Hs,5126 NM_001712 CEACAM1 Carcinoembryonic antigen-related cell adhesion molecule 1 1.31 26.08 25.59 B01 82Hs,2478 NM_005214 CTLA4 (biliaryCytotoxic glycoprotein) T-lymphocyte -associated protein 4 0.53 30.90 31.71 B11 24Hs,208 NM_000160 GCGR Glucagon receptor 0.93 35.00 35.00 C01 Hs,3891 NM_002062 GLP1R Glucagon-like peptide 1 receptor 0.93 35.00 35.00 C07 03Hs,6434 NM_000201 ICAM1 Intercellular adhesion molecule 1 0.84 28.74 28.89 D02 47Hs,5134 NM_000418 IL4R Interleukin 4 receptor 0.64 34.22 34.75 D06 57Hs,4657 NM_000208 INSR Insulin receptor 0.93 35.00 35.00 E05 44Hs,4312 NM_006178 NSF N-ethylmaleimide-sensitive factor 0.48 28.42 29.37 F08 79Hs,2961 NM_004578 RAB4A RAB4A, member RAS oncogene family 0.88 20.55 20.63 F10 69Hs,7287 NM_000655 SELL Selectin L 0.97 23.89 23.83 F11 56Hs,3806 NM_001042 SLC2A4 Solute carrier family 2 (facilitated glucose transporter), member 4 0.77 34.72 35.00 F12 91Hs,5111 NM_003825 SNAP23 Synaptosomal-associated protein, 23kDa 3.90 -

Nrf1 and Nrf2 Positively and C-Fos and Fra1 Negatively Regulate the Human Antioxidant Response Element-Mediated Expression of NAD(P)H:Quinone Oxidoreductase1 Gene
Proc. Natl. Acad. Sci. USA Vol. 93, pp. 14960–14965, December 1996 Pharmacology Nrf1 and Nrf2 positively and c-Fos and Fra1 negatively regulate the human antioxidant response element-mediated expression of NAD(P)H:quinone oxidoreductase1 gene RADJENDIRANE VENUGOPAL AND ANIL K. JAISWAL* Department of Pharmacology, Fox Chase Cancer Center, 7701 Burholme Avenue, Philadelphia, PA 19111 Communicated by Y. W. Kan, University of California, San Francisco, CA, August 5, 1996 (received for review February 21, 1996) ABSTRACT Twenty-four base pairs of the human anti- (c-Jun, Jun-D, and Jun-B) form homodimers among them- oxidant response element (hARE) are required for high basal selves and heterodimers with c-Fos and Fra (Fra1 and Fra2) transcription of the NAD(P)H:quinone oxidoreductase1 proteins and bind to the TRE (a single AP1 binding site) and (NQO1) gene and its induction in response to xenobiotics and regulate the expression of target genes (6). Nrf1 and Nrf2 are antioxidants. hARE is a unique cis-element that contains one recently identified NF-E2-related factors that bind to the perfect and one imperfect AP1 element arranged as inverse sequence GCTGAGTCATGATGAGTCA (7, 8). The Nrf1 repeats separated by 3 bp, followed by a ‘‘GC’’ box. We report and Nrf2 contain 742 and 589 amino acids, respectively. Both here that Jun, Fos, Fra, and Nrf nuclear transcription factors these proteins display marked homology to the NF-E2, which bind to the hARE. Overexpression of cDNA derived combi- regulates expression of globin genes in developing erythroid nations of the nuclear proteins Jun and Fos or Jun and Fra1 cells through interaction with upstream AP1-like recognition repressed hARE-mediated chloramphenicol acetyltransferase sites (9). -

Integrative Prediction of Gene Expression with Chromatin Accessibility and Conformation Data
bioRxiv preprint doi: https://doi.org/10.1101/704478; this version posted July 16, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Integrative prediction of gene expression with chromatin accessibility and conformation data Florian Schmidt1;2;3;y, Fabian Kern1;3;4;y, and Marcel H. Schulz1;2;3;5;6;∗ 1High-throughput Genomics & Systems Biology, Cluster of Excellence on Multimodal Computing and Interaction, Saarland Informatics Campus, 66123 Saarbr¨ucken Germany 2Computational Biology & Applied Algorithmics, Max-Planck Institute for Informatics, Saarland Informatics Campus, 66123 Saarbr¨ucken Germany 3Center for Bioinformatics, Saarland Informatics Campus, 66123 Saarbr¨ucken Germany 4Chair for Clinical Bioinformatics, Saarland Informatics Campus, 66123 Saarbr¨ucken Germany 5Institute of Cardiovascular Regeneration, Goethe-University, Theodor-Stern-Kai 7, 60590 Frankfurt am Main Germany 6German Center for Cardiovascular Research, Partner site Rhein-Main, Theodor-Stern-Kai 7, 60590 Frankfurt am Main Germany y These authors contributed equally ∗ Correspondence: [email protected] Abstract Background: Enhancers play a fundamental role in orchestrating cell state and development. Al- though several methods have been developed to identify enhancers, linking them to their target genes is still an open problem. Several theories have been proposed on the functional mechanisms of en- hancers, which triggered the development of various methods to infer promoter enhancer interactions (PEIs). The advancement of high-throughput techniques describing the three-dimensional organisa- tion of the chromatin, paved the way to pinpoint long-range PEIs. -

Transcription Factors Recognize DNA Shape Without Nucleotide Recognition
bioRxiv preprint doi: https://doi.org/10.1101/143677; this version posted May 29, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Transcription+factors+recognize+DNA+shape+without+nucleotide+recognition++ Md.$Abul$Hassan$Samee1,$Benoit$G.$Bruneau1,2,$Katherine$S.$Pollard$1,3$ 1:$Gladstone$Institutes,$1650$Owens$St.,$San$Francisco,$CA$94158$ 2:$$Department$of$Pediatrics$and$Cardiovascular$Research$Institute,$University$of$California,$ San$Francisco,$CA$94158$ 3:$Division$of$Bioinformatics,$Institute$for$Human$Genetics,$and$Institute$for$Computational$ Health$Sciences,$University$of$California,$San$Francisco,$CA$94158$ $ + bioRxiv preprint doi: https://doi.org/10.1101/143677; this version posted May 29, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract+ We$hypothesized$that$transcription$factors$(TFs)$recognize$DNA$shape$without$nucleotide$ sequence$recognition.$Motivating$an$independent$role$for$shape,$many$TF$binding$sites$lack$ a$sequenceZmotif,$DNA$shape$adds$specificity$to$sequenceZmotifs,$and$different$sequences$ can$ encode$ similar$ shapes.$ We$ therefore$ asked$ if$ binding$ sites$ of$ a$ TF$ are$ enriched$ for$ specific$patterns$of$DNA$shapeZfeatures,$e.g.,$helical$twist.$We$developed$ShapeMF,$which$ discovers$ these$ shapeZmotifs$ de%novo%without$ taking$ sequence$ information$ into$ account.$ -

Genetic Variation As a Tool for Identifying Novel Transducers of Itch
Genetic variation as a tool for identifying novel transducers of itch By Takeshi Morita A dissertation submitted in partial satisfaction of the requirements for the degree of Doctor of Philosophy in Molecular and Cell Biology in the Graduate Division of the University of California, Berkeley Committee in charge: Professor Diana M. Bautista, Co-Chair Professor Rachel B. Brem, Co-Chair Professor John Ngai Professor Kristin Scott Professor Michael W. Nachman Summer 2016 Abstract Genetic variation as a tool for identifying novel transducers of itch by Takeshi Morita Doctor of Philosophy in Molecular and Cell Biology University of California, Berkeley Professor Diana M. Bautista, Co-Chair Professor Rachel B. Brem, Co-Chair The mammalian somatosensory system mediates itch, the irritating sensation that elicits a desire to scratch. Millions of people worldwide suffer from chronic itch that fails to respond to current drugs and therapies. Even though recent studies have begun to elucidate the basic characteristics of the itch circuitry, we have little understanding about the molecules and signaling mechanisms that underlie detection and transduction of itch sensation, especially during chronic itch conditions. We have taken a genomic approach by harnessing natural variation in itch-evoked scratching behaviors in mice to identify novel molecular players that are involved in itch signal transduction at the level of primary sensory neurons. From our analysis, we identified numerous candidate itch genes, and further identified a serotonin receptor, HTR7 as a key transducer that is required for both development and maintenance of chronic itch. We further investigated the genetic basis of variation in itch, and identified a set of genes and regulatory pathways that may be involved in controlling itch behaviors.