Synthesis of 1,2,3,4-Tetrahydrocarbazoles with Large Groups - Aromatization to Carbazoles1 K
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
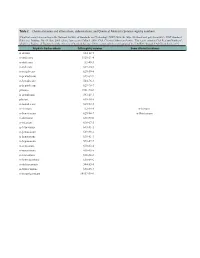
Table 2. Chemical Names and Alternatives, Abbreviations, and Chemical Abstracts Service Registry Numbers
Table 2. Chemical names and alternatives, abbreviations, and Chemical Abstracts Service registry numbers. [Final list compiled according to the National Institute of Standards and Technology (NIST) Web site (http://webbook.nist.gov/chemistry/); NIST Standard Reference Database No. 69, June 2005 release, last accessed May 9, 2008. CAS, Chemical Abstracts Service. This report contains CAS Registry Numbers®, which is a Registered Trademark of the American Chemical Society. CAS recommends the verification of the CASRNs through CAS Client ServicesSM] Aliphatic hydrocarbons CAS registry number Some alternative names n-decane 124-18-5 n-undecane 1120-21-4 n-dodecane 112-40-3 n-tridecane 629-50-5 n-tetradecane 629-59-4 n-pentadecane 629-62-9 n-hexadecane 544-76-3 n-heptadecane 629-78-7 pristane 1921-70-6 n-octadecane 593-45-3 phytane 638-36-8 n-nonadecane 629-92-5 n-eicosane 112-95-8 n-Icosane n-heneicosane 629-94-7 n-Henicosane n-docosane 629-97-0 n-tricosane 638-67-5 n-tetracosane 643-31-1 n-pentacosane 629-99-2 n-hexacosane 630-01-3 n-heptacosane 593-49-7 n-octacosane 630-02-4 n-nonacosane 630-03-5 n-triacontane 638-68-6 n-hentriacontane 630-04-6 n-dotriacontane 544-85-4 n-tritriacontane 630-05-7 n-tetratriacontane 14167-59-0 Table 2. Chemical names and alternatives, abbreviations, and Chemical Abstracts Service registry numbers.—Continued [Final list compiled according to the National Institute of Standards and Technology (NIST) Web site (http://webbook.nist.gov/chemistry/); NIST Standard Reference Database No. -

The Chemistry of Heterocycles
The Chemistry of Heterocycles Mohammad Jafarzadeh Faculty of Chemistry, Razi University The Chemistry of Heterocycles, (Second Edition). By Theophil Eicher and Siegfried Hauptmann, Wiley-VCH Veriag GmbH, 2003 1 110 5 Five-Membered Heterocycles The indole ring system forms the basis of several pharmaceuticals, for instance the anti-inflammatory indomethacin 54 and the antidepressant iprindole 55: MeO, I CH2— C H 2— C H 2— N M e 2 55 Indigo and other dyes (see p 81) which contain the chromophore 56 O x - ,X = N H o r S , ( E ) - o r ( Z ) - , 56 are known as indigoid dyes, and are vat dyes. These water-insoluble compounds are reduced by sodium dithionite and sodium hydroxide and applied as vat dyes, whereby they become soluble as disodium dihydro compounds, e.g.: ^2H,2NaOH -2H20 Before the availability of sodium dithionite (1871), the reduction of indigo was brought about by bacte- ria with reducing properties (fermentation vat). The vat of indigo has a brown-yellow colour. The cloth is dipped in the vat and then exposed to air to allow reoxidation to indigo which is precipitated and finely distributed onto the fibre. A consequence of this process is the low rubbing fastness of the dyes. It causes the faded appearance of indigo-dyed jeans and makes possible the manufacture of 'faded jeans'. Since the seventies, indole compound5.14s Isoindolehave lost their importance as textile dyes. They have, how- ever, found application in other fields, e.g. in polaroid photography. [A-C] The name isoindole is admissible for benzo[c]pyrrole5.15. -

Synthesis of Azaindoles, Diazaindoles, and Advanced Carbazole Alkaloid Intermediates Via Palladium-Catalyzed Reductive N- Heteroannulation
Graduate Theses, Dissertations, and Problem Reports 2008 Synthesis of azaindoles, diazaindoles, and advanced carbazole alkaloid intermediates via palladium-catalyzed reductive N- heteroannulation Grissell M. Carrero-Martinez West Virginia University Follow this and additional works at: https://researchrepository.wvu.edu/etd Recommended Citation Carrero-Martinez, Grissell M., "Synthesis of azaindoles, diazaindoles, and advanced carbazole alkaloid intermediates via palladium-catalyzed reductive N-heteroannulation" (2008). Graduate Theses, Dissertations, and Problem Reports. 4360. https://researchrepository.wvu.edu/etd/4360 This Thesis is protected by copyright and/or related rights. It has been brought to you by the The Research Repository @ WVU with permission from the rights-holder(s). You are free to use this Thesis in any way that is permitted by the copyright and related rights legislation that applies to your use. For other uses you must obtain permission from the rights-holder(s) directly, unless additional rights are indicated by a Creative Commons license in the record and/ or on the work itself. This Thesis has been accepted for inclusion in WVU Graduate Theses, Dissertations, and Problem Reports collection by an authorized administrator of The Research Repository @ WVU. For more information, please contact [email protected]. Synthesis of Azaindoles, Diazaindoles, and Advanced Carbazole Alkaloid Intermediates via Palladium-Catalyzed Reductive N-Heteroannulation Grissell M. Carrero-Martínez Thesis submitted to the Eberly College of Arts and Sciences at West Virginia University in partial fulfillment of the requirements for the degree of Master of Science in Chemistry Björn C. G. Söderberg, Ph.D., Chair Kung K. Wang, Ph.D. John H. Penn, Ph.D. -

Vinylation of a Secondary Amine Core with Calcium Carbide for Efficient
molecules Article Vinylation of a Secondary Amine Core with Calcium Carbide for Efficient Post-Modification and Access to Polymeric Materials Konstantin S. Rodygin 1, Alexander S. Bogachenkov 1 and Valentine P. Ananikov 1,2,* ID 1 Saint Petersburg State University, Universitetsky Prospect, 26, St. Petersburg 198504, Russia; [email protected] (K.S.R.); [email protected] (A.S.B.) 2 Zelinsky Institute of Organic Chemistry, Russian Academy of Science, Leninsky Prospect, 47, Moscow 119991, Russia * Correspondence: [email protected] Received: 16 February 2018; Accepted: 9 March 2018; Published: 13 March 2018 Abstract: We developed a simple and efficient strategy to access N-vinyl secondary amines of various naturally occurring materials using readily available solid acetylene reagents (calcium carbide, KF, and KOH). Pyrrole, pyrazole, indoles, carbazoles, and diarylamines were successfully vinylated in good yields. Cross-linked and linear polymers were synthesized from N-vinyl carbazoles through free radical and cationic polymerization. Post-modification of olanzapine (an antipsychotic drug substance) was successfully performed. Keywords: acetylene; calcium carbide; vinylation; olanzapine; functionalization 1. Introduction Nitrogen-containing compounds are one of the most important functional molecules in modern chemistry, biology, and drug development. Nucleic acids, N-heterocyclic compounds, amines, amino-acids, and their biopolymers are pivotal to a vast majority of biochemical processes. Effective modification of secondary amines by the insertion of a vinyl group leads to demanded N-vinyl derivatives with a variety of new applications. Vinylated products are interesting both as biologically active cores [1] and as versatile building blocks for polymerization [2]. At the same time, carbazole-based compounds are considered promising as photoconductors and charge-transporting materials [3] in the aftermath of the discovery of photoconductivity in poly(N-vinylcarbazole) [4]. -

Mutagens and Reproductive Toxins Chemical Class Standard Operating Procedure
1 Mutagens and Reproductive Toxins Chemical Class Standard Operating Procedure Mutagens and Reproductive Toxins H340 H341 H360 H361 H362 This SOP is not a substitute for hands-on training. Print a copy and insert into your laboratory SOP binder. Department: Chemistry Date SOP was written: Thursday, July 1, 2021 Date SOP was approved by PI/lab supervisor: Thursday, July 1, 2021 Name: F. Fischer Principal Investigator: Signature: ______________________________ Name: Matthew Rollings Internal Lab Safety Coordinator or Lab Manager: Lab Phone: 510.301.1058 Office Phone: 510.643.7205 Name: Felix Fischer Emergency Contact: Phone Number: 510.643.7205 Tan Hall 674, 675, 676, 679, 680, 683, 684 Location(s) covered by this SOP: Hildebrand Hall: D61, D32 1. Purpose This SOP covers the precautions and safe handling procedures for the use of Mutagens and Reproductive Toxins. For a list of Mutagens and Reproductive Toxins covered by this SOP and their use(s), see the “List of Chemicals”. Procedures described in Section 12 apply to all materials covered in this SOP. A change to the “List of Chemicals” does not constitute a change in the SOP requiring review or retraining. If you have questions concerning the applicability of any recommendation or requirement listed in this procedure, contact the Principal Investigator/Laboratory Supervisor or the campus Chemical Hygiene Officer at [email protected]. 2. Physical & Chemical Properties/Definition of Chemical Group Germ Cell Mutagenicity is a hazard class that is primarily concerned with chemicals that may cause mutations in the germ cell of humans that can be transmitted to the progeny. Rev. -

Influence of Electronic Environment on the Radiative Efficiency of 9
molecules Article Influence of Electronic Environment on the Radiative Efficiency of 9-Phenyl-9H-carbazole-Based ortho-Carboranyl Luminophores Seok Ho Lee †, Ji Hye Lee † , Min Sik Mun †, Sanghee Yi, Eunji Yoo, Hyonseok Hwang and Kang Mun Lee * Department of Chemistry, Institute for Molecular Science and Fusion Technology, Kangwon National University, Chuncheon 24341, Korea; [email protected] (S.H.L.); [email protected] (J.H.L.); [email protected] (M.S.M.); [email protected] (S.Y.); [email protected] (E.Y.); [email protected] (H.H.) * Correspondence: [email protected]; Tel.: +82-33-250-8499 † These authors contributed equally to the work. Abstract: The photophysical properties of closo-ortho-carboranyl-based donor–acceptor dyads are known to be affected by the electronic environment of the carborane cage but the influence of the elec- tronic environment of the donor moiety remains unclear. Herein, four 9-phenyl-9H-carbazole-based closo-ortho-carboranyl compounds (1F, 2P, 3M, and 4T), in which an o-carborane cage was appended at the C3-position of a 9-phenyl-9H-carbazole moiety bearing various functional groups, were syn- thesized and fully characterized using multinuclear nuclear magnetic resonance spectroscopy and elemental analysis. Furthermore, the solid-state molecular structures of 1F and 4T were determined by X-ray diffraction crystallography. For all the compounds, the lowest-energy absorption band Citation: Lee, S.H.; Lee, J.H.; exhibited a tail extending to 350 nm, attributable to the spin-allowed π–π* transition of the 9-phenyl- Mun, M.S.; Yi, S.; Yoo, E.; Hwang, H.; 9H-carbazole moiety and weak intramolecular charge transfer (ICT) between the o-carborane and Lee, K.M. -

Theoretical Study on the Acidities of Pyrrole, Indole, Carbazole and Their Hydrocarbon Analogues in DMSO
Canadian Journal of Chemistry Theoretical study on the acidities of pyrrole, indole, carbazole and their hydrocarbon analogues in DMSO Journal: Canadian Journal of Chemistry Manuscript ID cjc-2018-0032.R1 Manuscript Type: Article Date Submitted by the Author: 13-May-2018 Complete List of Authors: Ariche, Berkane; Modelisation and computational Méthods Laboratory, Faculty of Sciences, University of Saida, Chemistry RAHMOUNI,Draft Ali; Universite Dr Tahar Moulay de Saida, Chimie Is the invited manuscript for consideration in a Special Not applicable (regular submission) Issue?: Keyword: pKa calculation, Aromaticity index, Pyrolle, DMSO, DFT https://mc06.manuscriptcentral.com/cjc-pubs Page 1 of 33 Canadian Journal of Chemistry Theoretical study on the acidities of pyrrole, indole, carbazole and their hydrocarbon analogues in DMSO Ariche Berkane, Rahmouni Ali* Modeling and Calculation Methods Laboratory, University of Saida, B.P. 138, 20002 Saida, Algeria *Corresponding author [email protected] Abstract SMD and IEF-PCM continuum solvation models have been used, in combination with three quantum chemistry methods (B3LYP,Draft M062X and CBS-QB3) to study the acidities of pyrrole, indole and carbazole as well as their hydrocarbon analogues in DMSO, following a direct thermodynamic method. Theoretical parameters such as aromaticity indices (HOMA and SA), molecular electrostatic potential (MEP) and atomic charges have been calculated using B3LYP/6-311++G(d,p) level of theory. Calculated pKa values indicate that there is generally good agreement with experimental data, with all deviations being less than the acceptable error for a directly calculated pKa values. The M062X functional combined to SMD solvation model provide the more accurate pKa values. -

Synthesis of Carbazoles from N-(N,N-Diarylamino)Phthalimides with Aluminum Chloride Via Diarylnitrenium Ions
8612 J. Org. Chem. 2001, 66, 8612-8615 Synthesis of Carbazoles from N-(N,N-Diarylamino)phthalimides with Aluminum Chloride via Diarylnitrenium Ions Yasuo Kikugawa,* Yutaka Aoki, and Takeshi Sakamoto Faculty of Pharmaceutical Sciences, Josai University, 1-1 Keyakidai, Sakado, Saitama 350-0295, Japan [email protected] Received September 17, 2001 Generation of diarylnitrenium ions from N-(N,N-diarylamino)phthalimides by treatment with AlCl3 in benzene or in 1,2-dichloroethane leads to formation of carbazoles by intramolecular C-C bond formation. The reaction proceeds in synthetically useful yields. Introduction Scheme 1 From the extensive work of Gassman and his group, the nitrenium ion has received increasing attention from synthetic, theoretical, and biological perspectives.1 How- ever, despite the potential utility of the nitrenium ion as an electrophilic nitrogen, its synthetic applications re- main limited, mainly because it is a short-lived reaction intermediate. Previously, we reported that N-phenylaminophthal- imide reacts with AlCl3 in benzene to generate a phen- ylnitrenium ion that is trapped by benzene to give o- and p-aminobiphenyls and N-phenylaniline.2 On the other hand, with N-(N-methyl-N-phenylamino)phthalimide, the cleavage of the N-N bond results in a positive charge exclusively on the para position of the methylamino group, and 4-methylaminobiphenyl was obtained in 91% yield.3 In an extension of this work, we now have investigated the reaction of N-(N,N-diarylamino)phthal- imides (1) with AlCl3 in benzene, with the expectation that AlCl3-mediated cleavage of the N-N bond might give 4 a diarylnitrenium ion. -

Essentials of Heterocyclic Chemistry-I Heterocyclic Chemistry
Baran, Richter Essentials of Heterocyclic Chemistry-I Heterocyclic Chemistry 5 4 Deprotonation of N–H, Deprotonation of C–H, Deprotonation of Conjugate Acid 3 4 3 4 5 4 3 5 6 6 3 3 4 6 2 2 N 4 4 3 4 3 4 3 3 5 5 2 3 5 4 N HN 5 2 N N 7 2 7 N N 5 2 5 2 7 2 2 1 1 N NH H H 8 1 8 N 6 4 N 5 1 2 6 3 4 N 1 6 3 1 8 N 2-Pyrazoline Pyrazolidine H N 9 1 1 5 N 1 Quinazoline N 7 7 H Cinnoline 1 Pyrrolidine H 2 5 2 5 4 5 4 4 Isoindole 3H-Indole 6 Pyrazole N 3 4 Pyrimidine N pK : 11.3,44 Carbazole N 1 6 6 3 N 3 5 1 a N N 3 5 H 4 7 H pKa: 19.8, 35.9 N N pKa: 1.3 pKa: 19.9 8 3 Pyrrole 1 5 7 2 7 N 2 3 4 3 4 3 4 7 Indole 2 N 6 2 6 2 N N pK : 23.0, 39.5 2 8 1 8 1 N N a 6 pKa: 21.0, 38.1 1 1 2 5 2 5 2 5 6 N N 1 4 Pteridine 4 4 7 Phthalazine 1,2,4-Triazine 1,3,5-Triazine N 1 N 1 N 1 5 3 H N H H 3 5 pK : <0 pK : <0 3 5 Indoline H a a 3-Pyrroline 2H-Pyrrole 2-Pyrroline Indolizine 4 5 4 4 pKa: 4.9 2 6 N N 4 5 6 3 N 6 N 3 5 6 3 N 5 2 N 1 3 7 2 1 4 4 3 4 3 4 3 4 3 3 N 4 4 2 6 5 5 5 Pyrazine 7 2 6 Pyridazine 2 3 5 3 5 N 2 8 N 1 2 2 1 8 N 2 5 O 2 5 pKa: 0.6 H 1 1 N10 9 7 H pKa: 2.3 O 6 6 2 6 2 6 6 S Piperazine 1 O 1 O S 1 1 Quinoxaline 1H-Indazole 7 7 1 1 O1 7 Phenazine Furan Thiophene Benzofuran Isobenzofuran 2H-Pyran 4H-Pyran Benzo[b]thiophene Effects of Substitution on Pyridine Basicity: pKa: 35.6 pKa: 33.0 pKa: 33.2 pKa: 32.4 t 4 Me Bu NH2 NHAc OMe SMe Cl Ph vinyl CN NO2 CH(OH)2 4 8 5 4 9 1 3 2-position 6.0 5.8 6.9 4.1 3.3 3.6 0.7 4.5 4.8 –0.3 –2.6 3.8 6 3 3 5 7 4 8 2 3 5 2 3-position 5.7 5.9 6.1 4.5 4.9 4.4 2.8 4.8 4.8 1.4 0.6 3.8 4 2 6 7 7 3 N2 N 1 4-position -

Carbazole Violet Pigment 23 from China and India
Carbazole Violet Pigment 23 From China and India Investigations Nos. 701-TA-437 and 731-TA-1060 and 1061 (Final) Publication 3744 December 2004 U.S. International Trade Commission Washington, DC 20436 U.S. International Trade Commission COMMISSIONERS Stephen Koplan, Chairman Deanna Tanner Okun, Vice Chairman Marcia E. Miller Jennifer A. Hillman Charlotte R. Lane Daniel R. Pearson Robert A. Rogowsky Director of Operations Staff assigned Cynthia Trainor, Investigator Cynthia Foreso, Industry Analyst Craig Thomsen, Economist David Boyland, Accountant Mary Jane Alves, Attorney David Goldfine, Attorney Lemuel Shields, Statistician George Deyman, Supervisory Investigator Address all communications to Secretary to the Commission United States International Trade Commission Washington, DC 20436 U.S. International Trade Commission Washington, DC 20436 www.usitc.gov Carbazole Violet Pigment 23 From China and India Investigations Nos. 701-TA-437 and 731-TA-1060 and 1061 (Final) Publication 3744 December 2004 CONTENTS Page Determinations ................................................................. 1 Views of the Commission ......................................................... 3 Part I: Introduction ............................................................ I-1 Background .................................................................. I-1 Major firms involved in violet 23 market ........................................... I-2 Summary data................................................................ I-2 Nature and extent of subsidies -

Synthesis and Characterization of New Conjugated Polymers for Application in Solar Cells
Synthesis and Characterization of New Conjugated Polymers for Application in Solar Cells Mohd Sani Bin Sarjadi A thesis submitted to The University of Sheffield as partial fulfillment for the degree of Doctor of Philosophy October 2014 Declaration This thesis is submitted for the degree of doctorate of philosophy (PhD) at the University of Sheffield, having been submitted for no other degree. It records the research carried out the University of Sheffield from November 2009 to October 2014. It is entirely my original work, unless where referenced. Signed:…………………………………………………….. 15 OCTOBER 2014 Date:…………………………………………………… i Abstract Plastic photovoltaic devices based on solution processed conjugated polymers have attracted much attention as potential candidates for the next generation of solar cells. Polymer solar cells based on blends of conjugated polymer donors and molecular acceptors such as PCBM (often referred to as bulk heterojunction solar cells) are attracting a great deal of interest. These systems have potential technological value due to their ease of fabrication and their relatively low production costs. This enabled devices to be produced that have solar power conversion efficiencies approaching 9%. The purpose of this research project is to develop new and more efficient materials for application in this area. A new class of alternating copolymers comprising carbazole units and thieno‐thiophene units for application in the area of bulk heterojunction solar cells is presented in this contribution. The polymers were prepared using Suzuki coupling methods. Three main classes of such polymers were prepared. The first class of polymers P1 ‐ P3 consisted of alternating co‐ polymers comprising thienothiophene repeat units and either 2,7‐linked 9‐alkyl carbazole units P1, or 2,7‐linked‐9‐alkyl carbazole repeat units with fluorine substituents at their 3,6‐ positions P2, or 2,7‐linked‐9,9‐dioctylfluorene P3. -

Some Correlations of Carbazole Derivatives with Related Heterocycles Julian Bernard Honeycutt Iowa State College
Iowa State University Capstones, Theses and Retrospective Theses and Dissertations Dissertations 1956 Some correlations of carbazole derivatives with related heterocycles Julian Bernard Honeycutt Iowa State College Follow this and additional works at: https://lib.dr.iastate.edu/rtd Part of the Organic Chemistry Commons Recommended Citation Honeycutt, Julian Bernard, "Some correlations of carbazole derivatives with related heterocycles " (1956). Retrospective Theses and Dissertations. 13972. https://lib.dr.iastate.edu/rtd/13972 This Dissertation is brought to you for free and open access by the Iowa State University Capstones, Theses and Dissertations at Iowa State University Digital Repository. It has been accepted for inclusion in Retrospective Theses and Dissertations by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. NOTE TO USERS This reproduction is the best copy available. UMI SOME CORRPXATIOMS OF CAREAZOLE DERIVATIVES WITH RELATED HETElloCYCLES by Julian Bernard Honeycutt, Jr A Dleeertatlon Submitted to the Graduate Faculty In Partial Fulfillment of The Requirements for the Degree of DOCTOR OF PHILOBOPHT Major Subject: Organic Chemistry Approved: Signature was redacted for privacy. In Charge of Major Work Signature was redacted for privacy. Head of Major Department^' Signature was redacted for privacy. Dean of/G-raduate College Iowa State College 1956 UMI Number: DP12765 INFORMATION TO USERS The quality of this reproduction is dependent upon the quality of the copy submitted. Broken or indistinct print, colored or poor quality illustrations and photographs, print bleed-through, substandard margins, and improper alignment can adversely affect reproduction. In the unlikely event that the author did not send a complete manuscript and there are missing pages, these will be noted.