Protocol Template with Guidelines
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

2021 – the Following CPT Codes Are Approved for Billing Through Women’S Way
WHAT’S COVERED – 2021 Women’s Way CPT Code Medicare Part B Rate List Effective January 1, 2021 For questions, call the Women’s Way State Office 800-280-5512 or 701-328-2389 • CPT codes that are specifically not covered are 77061, 77062 and 87623 • Reimbursement for treatment services is not allowed. (See note on page 8). • CPT code 99201 has been removed from What’s Covered List • New CPT codes are in bold font. 2021 – The following CPT codes are approved for billing through Women’s Way. Description of Services CPT $ Rate Office Visits New patient; medically appropriate history/exam; straightforward decision making; 15-29 minutes 99202 72.19 New patient; medically appropriate history/exam; low level decision making; 30-44 minutes 99203 110.77 New patient; medically appropriate history/exam; moderate level decision making; 45-59 minutes 99204 165.36 New patient; medically appropriate history/exam; high level decision making; 60-74 minutes. 99205 218.21 Established patient; evaluation and management, may not require presence of physician; 99211 22.83 presenting problems are minimal Established patient; medically appropriate history/exam, straightforward decision making; 10-19 99212 55.88 minutes Established patient; medically appropriate history/exam, low level decision making; 20-29 minutes 99213 90.48 Established patient; medically appropriate history/exam, moderate level decision making; 30-39 99214 128.42 minutes Established patient; comprehensive history exam, high complex decision making; 40-54 minutes 99215 128.42 Initial comprehensive -

Agreement Between Endocervical Brush and Endocervical Curettage in Patients Undergoing Repeat Endocervical Sampling
Agreement Between Endocervical Brush and Endocervical Curettage in Patients Undergoing Repeat Endocervical Sampling Meredith J. Alston MD David W. Doo MD Sara E. Mazzoni MD MPH Elaine H. Stickrath MD Denver Health Medical Center University of Colorado Department of Obstetrics and Gynecology Background • Women with abnormal Pap tests referred for colposcopy frequently require sampling of the endocervix • ASCCP deems both the endocervical brush (ECB) and the endocervical currette (ECC) appropriate means of collecting endocervical samples (1) • Both have advantages and disadvantages, and it is unclear if one modality is superior Background • ECB has good sensitivity for endocervical lesions, it has poor specificity, ranging from 26- 38%2,3 • ECC has an excellent negative predictive value of 99.4% in women who had a satisfactory colposcopy • ECB is better tolerated by the patient, but runs the risk of contamination from the ectocervix • ECC is less likely to obtain an adequate sample Background • The results from the endocervical sample may influence clinical management after colposcopy. • Certain treatment options for high grade squamous intraepithelial neoplasia are available only to women with negative endocervical sampling. ▫ Ablative techniques ▫ Expectant management Background • Over concerns related to potential complications of excisional treatments, as well as over- treatment, there has been a push to re-introduce ablative techniques and, in appropriate clinical circumstances, expectant management, into the routine treatment of patients with cervical cancer precursors (12). Background • At our institution, it is our concern that due to the possibility of contamination from the ectocervix, a positive ECB may not represent a true positive endocervical sample. • We do not use a sleeve • Positive ECBs return for ECC if otherwise a candidate for ablative therapy or expectant management Objective • To describe the agreement between these two modalities of endocervical sampling. -
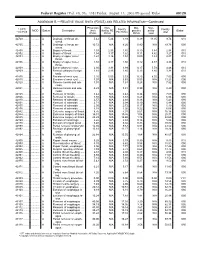
RELATIVE VALUE UNITS (RVUS) and RELATED INFORMATION—Continued
Federal Register / Vol. 68, No. 158 / Friday, August 15, 2003 / Proposed Rules 49129 ADDENDUM B.—RELATIVE VALUE UNITS (RVUS) AND RELATED INFORMATION—Continued Physician Non- Mal- Non- 1 CPT/ Facility Facility 2 MOD Status Description work facility PE practice acility Global HCPCS RVUs RVUs PE RVUs RVUs total total 42720 ....... ........... A Drainage of throat ab- 5.42 5.24 3.93 0.39 11.05 9.74 010 scess. 42725 ....... ........... A Drainage of throat ab- 10.72 N/A 8.26 0.80 N/A 19.78 090 scess. 42800 ....... ........... A Biopsy of throat ................ 1.39 2.35 1.45 0.10 3.84 2.94 010 42802 ....... ........... A Biopsy of throat ................ 1.54 3.17 1.62 0.11 4.82 3.27 010 42804 ....... ........... A Biopsy of upper nose/ 1.24 3.16 1.54 0.09 4.49 2.87 010 throat. 42806 ....... ........... A Biopsy of upper nose/ 1.58 3.17 1.66 0.12 4.87 3.36 010 throat. 42808 ....... ........... A Excise pharynx lesion ...... 2.30 3.31 1.99 0.17 5.78 4.46 010 42809 ....... ........... A Remove pharynx foreign 1.81 2.46 1.40 0.13 4.40 3.34 010 body. 42810 ....... ........... A Excision of neck cyst ........ 3.25 5.05 3.53 0.25 8.55 7.03 090 42815 ....... ........... A Excision of neck cyst ........ 7.07 N/A 5.63 0.53 N/A 13.23 090 42820 ....... ........... A Remove tonsils and ade- 3.91 N/A 3.63 0.28 N/A 7.82 090 noids. -
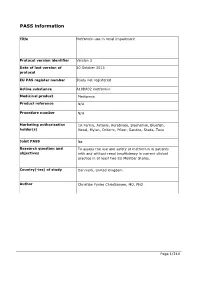
Guidance for the Format and Content of the Protocol of Non-Interventional
PASS information Title Metformin use in renal impairment Protocol version identifier Version 2 Date of last version of 30 October 2013 protocol EU PAS register number Study not registered Active substance A10BA02 metformin Medicinal product Metformin Product reference N/A Procedure number N/A Marketing authorisation 1A Farma, Actavis, Aurobindo, Biochemie, Bluefish, holder(s) Hexal, Mylan, Orifarm, Pfizer, Sandoz, Stada, Teva Joint PASS No Research question and To assess the use and safety of metformin in patients objectives with and without renal insufficiency in current clinical practice in at least two EU Member States. Country(-ies) of study Denmark, United Kingdom Author Christian Fynbo Christiansen, MD, PhD Page 1/214 Marketing authorisation holder(s) Marketing authorisation N/A holder(s) MAH contact person N/A Page 2/214 1. Table of Contents PASS information .......................................................................................................... 1 Marketing authorisation holder(s) .................................................................................... 2 1. Table of Contents ...................................................................................................... 3 2. List of abbreviations ................................................................................................... 4 3. Responsible parties .................................................................................................... 5 4. Abstract .................................................................................................................. -

Obstetrics and Gyneclogy
3/28/2016 Obstetrics and Gynecology Presented by: Peggy Stilley, CPC, CPC-I, CPMA, CPB, COBGC Objectives • Procedures • Pregnancy • Payments • Patient Relationships 1 3/28/2016 Female Genital Anatomy Terminology and Abbreviations • Endometriosis • Neoplasm • BUS • TAH/BSO • G3P2 2 3/28/2016 Procedures • Hysterectomy • Prolapse repairs • IUDs • Colposcopy Hysterectomy • Approach • Open • Vaginal • Total Laparoscopic • Laparoscopic assisted • Extent • Total • Subtotal • Supracervical • Diagnosis 3 3/28/2016 CPT Codes • Abdominal 58150 – • With or without removal tubes/ovaries 58240 • Some additional services • Vaginal 58260-58270 • Size of uterus < 250 grams, > 250 grams 58275-58294 • Additional services CPT Codes • LAVH 58541-58544 • Detach uterus , cervix, and structures through the scope 58548-58554 • Uterus removed thru the vagina • TLH • Detach structures laparoscopically entire 58570- 58573 uterus, cervix, bodies • Removed thru the vagina or abdomen • LSH • Detaching structures through the scope, 58541 – 58544 leaving the cervix • Morcellating – removing abdominally 4 3/28/2016 Hysterectomy Additional procedures performed • Tubes & Ovaries removed • Enterocele repair • Repairs for incontinence • Marshall-Marchetti-Krantz • Colporrhaphy • Colpo-urethropexy • Urethral Sling • TVT, TOT 5 3/28/2016 Procedures • 57288 Sling • 57240 Anterior Repair • 57250 Posterior Repair • +57267 Add on code for mesh/graft • 57260 Combo of A&P • 57425 Laparoscopic Colpopexy • 57280 Colpopexy, Abdominal approach • 57282 Colpopexy, vaginal approach Example 1 PREOPERATIVE DIAGNOSES: 1. Menorrhagia unresponsive to medical treatment with resulting chronic blood loss anemia POSTOPERATIVE DIAGNOSES: 1. Menorrhagia 2. Blood loss anemia TITLE OF SURGERY: Total abdominal hysterectomy ANESTHESIA: GENERAL ENDOTRACHEAL ANESTHESIA. INDICATIONS: The patient is a lovely 52-year-old female who presented with menorrhagia that is non- responsive to medical treatment. -

Tnm Frequently Asked Questions (Faq’S)
TNM FREQUENTLY ASKED QUESTIONS (FAQ’S) The TNM Project Committee receives questions concerning the use of TNM and how to interpret rules in specific situations. Some questions and answers are listed below by category for your convenience. These FAQs can th also be found in the TNM Supplement: a Commentary on Uniform Use, 4 Edition, 2012 (edited by Ch. Wittekind, C. Compton, J. Brierley, L. H. Sobin). Advice on further questions may be obtained from the TNM Helpdesk by accessing the TNM Classification of Malignant Tumours page at the UICC website www.uicc.org TABLE OF CONTENTS GENERAL QUESTIONS AJCC versus UICC TNM ................................................................................................................3 In situ carcinoma .............................................................................................................................3 Pathological Versus Clinical TNM ...................................................................................................3 When in Doubt ................................................................................................................................4 R Classification ...............................................................................................................................4 R Classification and Tis ..................................................................................................................5 Positive Cytology ............................................................................................................................5 -

2019 Reimbursement Schedule IMPORTANT INFORMATION REGARDING REIMBURSEMENT by the CARE for YOURSELF PROGRAM 1
Iowa Department of Public Health Effective on or after Date of Service 01.01.2019 Division of Health Promotion Chronic Disease Prevention Iowa Care for Yourself Program 2019 Reimbursement Schedule IMPORTANT INFORMATION REGARDING REIMBURSEMENT BY THE CARE FOR YOURSELF PROGRAM 1. If a Pap test is performed, the collection of the Pap (CPT codes 99000, Q0091 & Q0111) is included in the office visit reimbursement. The woman is not to be billed for the collection or handling of the specimen. 2. These amounts apply when service is performed for the purpose of this program. Rates listed for services include all incidental charges related to the procedure; additional amounts may not be billed to the client. 3. Federal funding can not be used to reimburse for treatment of breast cancer, cervical intraepithelial neoplasia or cervical cancer. CPT Description End RATE Code OFFICE VISITS Notes 26 TC Total 99201 New Patient Visit; problem focused 3 42.90 99202 New Patient Visit; expanded problem focused 3 72.00 99203 New Patient Visit; detailed, low complexity 3 102.01 99204 New Patient Visit; comprehensive history, exam, moderate complexity 1 155.82 99205 New Patient Visit; comprehensive history, exam, high complexity 1,3 196.16 99211 Established Patient Visit, may not require presence of physician 21.35 99212 Established Patient Visit, problem focused 3,4 42.42 99213 Established Patient Visit, expanded problem focused 3,4 70.35 99214 Established Patient Visit, comprehensive moderate complexity 3,4 103.31 99215 Established Patient Visit, comprehensive high complexity 3,4 138.47 99385 New Patient Visit (18 - 39 y.o) - paid at 99203 rate 2 102.01 99386 New Patient Visit (40 - 64 y.o.) - paid at 99203 rate 2 102.01 99387 New Patient Visit (65+ y.o.) - paid at 99203 rate 2 102.01 99395 Established Patient Visit (18 - 39 y.o.) - paid at 99213 rate 2 70.35 99396 Established Patient Visit (40 - 64 y.o.) - paid at 99213 rate 2,3,4 70.35 99397 Established Patient Visit (65+ y.o.) - paid at 99213 rate 2 70.35 G0101 Cancer screening; pelvic and breast exam included. -

Ministry of Education and Science of Ukraine Sumy State University 0
Ministry of Education and Science of Ukraine Sumy State University 0 Ministry of Education and Science of Ukraine Sumy State University SPLANCHNOLOGY, CARDIOVASCULAR AND IMMUNE SYSTEMS STUDY GUIDE Recommended by the Academic Council of Sumy State University Sumy Sumy State University 2016 1 УДК 611.1/.6+612.1+612.017.1](072) ББК 28.863.5я73 С72 Composite authors: V. I. Bumeister, Doctor of Biological Sciences, Professor; L. G. Sulim, Senior Lecturer; O. O. Prykhodko, Candidate of Medical Sciences, Assistant; O. S. Yarmolenko, Candidate of Medical Sciences, Assistant Reviewers: I. L. Kolisnyk – Associate Professor Ph. D., Kharkiv National Medical University; M. V. Pogorelov – Doctor of Medical Sciences, Sumy State University Recommended for publication by Academic Council of Sumy State University as а study guide (minutes № 5 of 10.11.2016) Splanchnology Cardiovascular and Immune Systems : study guide / С72 V. I. Bumeister, L. G. Sulim, O. O. Prykhodko, O. S. Yarmolenko. – Sumy : Sumy State University, 2016. – 253 p. This manual is intended for the students of medical higher educational institutions of IV accreditation level who study Human Anatomy in the English language. Посібник рекомендований для студентів вищих медичних навчальних закладів IV рівня акредитації, які вивчають анатомію людини англійською мовою. УДК 611.1/.6+612.1+612.017.1](072) ББК 28.863.5я73 © Bumeister V. I., Sulim L G., Prykhodko О. O., Yarmolenko O. S., 2016 © Sumy State University, 2016 2 Hippocratic Oath «Ὄμνυμι Ἀπόλλωνα ἰητρὸν, καὶ Ἀσκληπιὸν, καὶ Ὑγείαν, καὶ Πανάκειαν, καὶ θεοὺς πάντας τε καὶ πάσας, ἵστορας ποιεύμενος, ἐπιτελέα ποιήσειν κατὰ δύναμιν καὶ κρίσιν ἐμὴν ὅρκον τόνδε καὶ ξυγγραφὴν τήνδε. -

2012 Updated Consensus Guidelines for Managing Abnormal Cervical
2012 Updated Consensus Guidelines for the Management of Abnormal Cervical Cancer Screening Tests and Cancer Precursors L. Stewart Massad, MD, Mark H. Einstein, MD, Warner K. Huh, MD, Hormuzd A. Katki, PhD, Walter K. Kinney, MD, Mark Schiffman, MD, Diane Solomon, MD, Nicolas Wentzensen, MD, and Herschel W. Lawson, MD, for the 2012 ASCCP Consensus Guidelines Conference From Washington University School of Medicine, St. Louis, Missouri; Albert Einstein College of Medicine, New York, New York; University of Alabama School of Medicine, Birmingham, Alabama; Division of Cancer Epidemiology and Genetics and Division of Cancer Prevention, National Cancer Institute, Bethesda, Maryland; The Permanente Medical Group, Sacramento, California; and Emory University School of Medicine, Atlanta, Georgia h ABSTRACT: A group of 47 experts representing 23 Kaiser Permanente Northern California Medical Care Plan professional societies, national and international health provided evidence on risk after abnormal tests. Where data organizations, and federal agencies met in Bethesda, MD, were available, guidelines prescribed similar management September 14Y15, 2012, to revise the 2006 American Society for women with similar risks forCIN3,AIS,andcancer.Most for Colposcopy and Cervical Pathology Consensus Guidelines. prior guidelines were reaffirmed. Examples of updates in- The group’s goal was to provide revised evidence-based clude: Human papillomavirusYnegative atypical squamous consensus guidelines for managing women with abnormal cells of undetermined significance results are followed with cervical cancer screening tests, cervical intraepithelial neo- co-testing at 3 years before return to routine screening and plasia (CIN) and adenocarcinoma in situ (AIS) following are not sufficient for exiting women from screening at age adoption of cervical cancer screening guidelines incorporat- 65 years; women aged 21Y24 years need less invasive man- ing longer screening intervals and co-testing. -
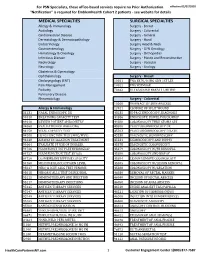
Services Not Requiring Prior Authorization for PSN Specialists
For PSN Specialists, these office-based services require no Prior Authorization. effective 02/07/2020 "Notification" is required for EmblemHealth Cohort 2 patients - see website for details MEDICAL SPECIALTIES SURGICAL SPECIALTIES Allergy & Immunology Surgery - Breast Audiology Surgery - Colorectal Cardiovascular Disease Surgery - General Dermatology & Dermatopathology Surgery - Hand Endocrinology Surgery Head & Neck Gastroenterology Surgery - GYN Oncology Hematology & Oncology Surgery - Orthopedics Infectious Disease Surgery - Plastic and Reconstructive Nephrology Surgery - Vascular Neurology Surgery - Urology Obstetrics & Gynecology Ophthalmology Surgery - Breast Otolaryngology (ENT) 10021 FNA BX W/O IMG GDN 1ST LES Pain Management 10022 FNA W/IMAGE Podiatry 76642 ULTRASOUND BREAST LIMITED Pulmonary Disease Rheumatology Surgery - Colorectal 10060 DRAINAGE OF SKIN ABSCESS Allergy & Immunology 12021 CLOSURE OF SPLIT WOUND 31231 NASAL ENDOSCOPY, DX 43235 UPPR GI ENDOSCOPY, DIAGNOSIS 94010 BREATHING CAPACITY TEST 44386 ENDOSCOPY, BOWEL POUCH/BIOP 94016 REVIEW PATIENT SPIROMETRY 44388 COLONOSCOPY THRU STOMA SPX 94060 EVALUATION OF WHEEZING 45300 PROCTOSIGMOIDOSCOPY DX 94150 VITAL CAPACITY TEST 45303 PROCTOSIGMOIDOSCOPY DILATE 94200 LUNG FUNCTION TEST (MBC/MVV) 45330 DIAGNOSTIC SIGMOIDOSCOPY 94640 AIRWAY INHALATION TREATMENT 45331 SIGMOIDOSCOPY AND BIOPSY 94664 EVALUATE PT USE OF INHALER 45378 DIAGNOSTIC COLONOSCOPY 94726 PULM FUNCT TST PLETHYSMOGRAP 45379 COLONOSCOPY W/FB REMOVAL 94727 PULM FUNCTION TEST BY GAS 45380 COLONOSCOPY AND BIOPSY -

SJH Procedures
SJH Procedures - Gynecology and Gynecology Oncology Services New Name Old Name CPT Code Service ABLATION, LESION, CERVIX AND VULVA, USING CO2 LASER LASER VAPORIZATION CERVIX/VULVA W CO2 LASER 56501 Destruction of lesion(s), vulva; simple (eg, laser surgery, Gynecology electrosurgery, cryosurgery, chemosurgery) 56515 Destruction of lesion(s), vulva; extensive (eg, laser surgery, Gynecology electrosurgery, cryosurgery, chemosurgery) 57513 Cautery of cervix; laser ablation Gynecology BIOPSY OR EXCISION, LESION, FACE AND NECK EXCISION/BIOPSY (MASS/LESION/LIPOMA/CYST) FACE/NECK General, Gynecology, Plastics, ENT, Maxillofacial BIOPSY OR EXCISION, LESION, FACE AND NECK, 2 OR MORE EXCISE/BIOPSY (MASS/LESION/LIPOMA/CYST) MULTIPLE FACE/NECK 11102 Tangential biopsy of skin (eg, shave, scoop, saucerize, curette); General, Gynecology, single lesion Aesthetics, Urology, Maxillofacial, ENT, Thoracic, Vascular, Cardiovascular, Plastics, Orthopedics 11103 Tangential biopsy of skin (eg, shave, scoop, saucerize, curette); General, Gynecology, each separate/additional lesion (list separately in addition to Aesthetics, Urology, code for primary procedure) Maxillofacial, ENT, Thoracic, Vascular, Cardiovascular, Plastics, Orthopedics 11104 Punch biopsy of skin (including simple closure, when General, Gynecology, performed); single lesion Aesthetics, Urology, Maxillofacial, ENT, Thoracic, Vascular, Cardiovascular, Plastics, Orthopedics 11105 Punch biopsy of skin (including simple closure, when General, Gynecology, performed); each separate/additional lesion -

Cervical Cancer
Cervical Cancer Overview Each year, about 15,000 women in the United States learn they have cervical cancer. Cervical cancer is one of the most common types to affect a woman’s reproductive organs. Most cases of cervical cancer are caused by the human papillomavirus (HPV). Before cervical cancer appears, the cells of the cervix go through precancerous changes known as dysplasia. It is these abnormal cell changes that an annual Pap test can help to detect. For some women, these changes may go away without any treatment. More often, they need to be treated to keep them from changing into cancer. Because so many women do have Pap tests annually, deaths from cervical cancer have decreased greatly and are now rare in the United States. Chances of successfully treating cervical cancer are highest when detected early. Need-to-Know Anatomy The cervix is part of the female reproductive system. It is the narrow, lower end of the uterus. It is a hollow, pear-shaped organ where a fetus grows. It is about one inch around and connects the vagina (birth canal) to the uterus. During a woman’s menstrual period, blood flows from the uterus through the cervix to the vagina. The cervix also produces mucus that helps sperm move from the vagina into the uterus. Sperm travel through the cervix to fertilize a woman’s egg during conception. During pregnancy, the cervix is tightly closed to help keep the baby inside the uterus. During childbirth, the cervix opens or dilates to allow the baby to pass through the vagina.