Integrative Analysis of Genomic Amplification-Dependent Expression
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Whole-Genome Microarray Detects Deletions and Loss of Heterozygosity of Chromosome 3 Occurring Exclusively in Metastasizing Uveal Melanoma
Anatomy and Pathology Whole-Genome Microarray Detects Deletions and Loss of Heterozygosity of Chromosome 3 Occurring Exclusively in Metastasizing Uveal Melanoma Sarah L. Lake,1 Sarah E. Coupland,1 Azzam F. G. Taktak,2 and Bertil E. Damato3 PURPOSE. To detect deletions and loss of heterozygosity of disease is fatal in 92% of patients within 2 years of diagnosis. chromosome 3 in a rare subset of fatal, disomy 3 uveal mela- Clinical and histopathologic risk factors for UM metastasis noma (UM), undetectable by fluorescence in situ hybridization include large basal tumor diameter (LBD), ciliary body involve- (FISH). ment, epithelioid cytomorphology, extracellular matrix peri- ϩ ETHODS odic acid-Schiff-positive (PAS ) loops, and high mitotic M . Multiplex ligation-dependent probe amplification 3,4 5 (MLPA) with the P027 UM assay was performed on formalin- count. Prescher et al. showed that a nonrandom genetic fixed, paraffin-embedded (FFPE) whole tumor sections from 19 change, monosomy 3, correlates strongly with metastatic death, and the correlation has since been confirmed by several disomy 3 metastasizing UMs. Whole-genome microarray analy- 3,6–10 ses using a single-nucleotide polymorphism microarray (aSNP) groups. Consequently, fluorescence in situ hybridization were performed on frozen tissue samples from four fatal dis- (FISH) detection of chromosome 3 using a centromeric probe omy 3 metastasizing UMs and three disomy 3 tumors with Ͼ5 became routine practice for UM prognostication; however, 5% years’ metastasis-free survival. to 20% of disomy 3 UM patients unexpectedly develop metas- tases.11 Attempts have therefore been made to identify the RESULTS. Two metastasizing UMs that had been classified as minimal region(s) of deletion on chromosome 3.12–15 Despite disomy 3 by FISH analysis of a small tumor sample were found these studies, little progress has been made in defining the key on MLPA analysis to show monosomy 3. -

Analysis of Gene Expression Data for Gene Ontology
ANALYSIS OF GENE EXPRESSION DATA FOR GENE ONTOLOGY BASED PROTEIN FUNCTION PREDICTION A Thesis Presented to The Graduate Faculty of The University of Akron In Partial Fulfillment of the Requirements for the Degree Master of Science Robert Daniel Macholan May 2011 ANALYSIS OF GENE EXPRESSION DATA FOR GENE ONTOLOGY BASED PROTEIN FUNCTION PREDICTION Robert Daniel Macholan Thesis Approved: Accepted: _______________________________ _______________________________ Advisor Department Chair Dr. Zhong-Hui Duan Dr. Chien-Chung Chan _______________________________ _______________________________ Committee Member Dean of the College Dr. Chien-Chung Chan Dr. Chand K. Midha _______________________________ _______________________________ Committee Member Dean of the Graduate School Dr. Yingcai Xiao Dr. George R. Newkome _______________________________ Date ii ABSTRACT A tremendous increase in genomic data has encouraged biologists to turn to bioinformatics in order to assist in its interpretation and processing. One of the present challenges that need to be overcome in order to understand this data more completely is the development of a reliable method to accurately predict the function of a protein from its genomic information. This study focuses on developing an effective algorithm for protein function prediction. The algorithm is based on proteins that have similar expression patterns. The similarity of the expression data is determined using a novel measure, the slope matrix. The slope matrix introduces a normalized method for the comparison of expression levels throughout a proteome. The algorithm is tested using real microarray gene expression data. Their functions are characterized using gene ontology annotations. The results of the case study indicate the protein function prediction algorithm developed is comparable to the prediction algorithms that are based on the annotations of homologous proteins. -

1 Supporting Information for a Microrna Network Regulates
Supporting Information for A microRNA Network Regulates Expression and Biosynthesis of CFTR and CFTR-ΔF508 Shyam Ramachandrana,b, Philip H. Karpc, Peng Jiangc, Lynda S. Ostedgaardc, Amy E. Walza, John T. Fishere, Shaf Keshavjeeh, Kim A. Lennoxi, Ashley M. Jacobii, Scott D. Rosei, Mark A. Behlkei, Michael J. Welshb,c,d,g, Yi Xingb,c,f, Paul B. McCray Jr.a,b,c Author Affiliations: Department of Pediatricsa, Interdisciplinary Program in Geneticsb, Departments of Internal Medicinec, Molecular Physiology and Biophysicsd, Anatomy and Cell Biologye, Biomedical Engineeringf, Howard Hughes Medical Instituteg, Carver College of Medicine, University of Iowa, Iowa City, IA-52242 Division of Thoracic Surgeryh, Toronto General Hospital, University Health Network, University of Toronto, Toronto, Canada-M5G 2C4 Integrated DNA Technologiesi, Coralville, IA-52241 To whom correspondence should be addressed: Email: [email protected] (M.J.W.); yi- [email protected] (Y.X.); Email: [email protected] (P.B.M.) This PDF file includes: Materials and Methods References Fig. S1. miR-138 regulates SIN3A in a dose-dependent and site-specific manner. Fig. S2. miR-138 regulates endogenous SIN3A protein expression. Fig. S3. miR-138 regulates endogenous CFTR protein expression in Calu-3 cells. Fig. S4. miR-138 regulates endogenous CFTR protein expression in primary human airway epithelia. Fig. S5. miR-138 regulates CFTR expression in HeLa cells. Fig. S6. miR-138 regulates CFTR expression in HEK293T cells. Fig. S7. HeLa cells exhibit CFTR channel activity. Fig. S8. miR-138 improves CFTR processing. Fig. S9. miR-138 improves CFTR-ΔF508 processing. Fig. S10. SIN3A inhibition yields partial rescue of Cl- transport in CF epithelia. -

AIFM2 Monoclonal Antibody (M13A), Clone 2C6
AIFM2 monoclonal antibody (M13A), clone 2C6 Catalog # : H00084883-M13A 規格 : [ 200 uL ] List All Specification Application Image Product Mouse monoclonal antibody raised against a partial recombinant Western Blot (Transfected lysate) Description: AIFM2. Immunogen: AIFM2 (AAH06121, 1 a.a. ~ 339 a.a) partial recombinant protein with GST tag. MW of the GST tag alone is 26 KDa. Sequence: MGSQVSVESGALHVVIVGGGFGGIAAASQLQALNVPFMLVDMKDSFHHN VAALRASVETGFAKKTFISYSVTFKDNFRQGLVVGIDLKNQMVLLQGGE ALPFSHLILATGSTGPFPGKFNEVSSQQAAIQAYEDMVRQVQRSRFIVVV enlarge GGGSAGVEMAAEIKTEYPEKEVTLIHSQVALADKELLPSVRQEVKEILLRK Western Blot (Recombinant GVQLLLSERVSNLEELPLNEYREYIKVQTDKGTEVATNLVILCTGIKINSSA protein) YRKAFESRLASSGALRVNEHLQVEGHSNVYAIGDCADVRTPKMAYLAGL HANIAVANIVNSVKQRPLQAYKPGALTFLLSMGRNDGVG ELISA Host: Mouse Reactivity: Human Isotype: IgG1 Kappa Quality Control Antibody Reactive Against Recombinant Protein. Testing: Western Blot detection against Immunogen (62.92 KDa) . Storage Buffer: In ascites fluid Storage Store at -20°C or lower. Aliquot to avoid repeated freezing and thawing. Instruction: MSDS: Download Interspecies Mouse (90); Rat (90) Antigen Sequence: Datasheet: Download Applications Western Blot (Transfected lysate) Page 1 of 3 2021/6/18 Western Blot analysis of AIFM2 expression in transfected 293T cell line by AMID monoclonal antibody (M13A), clone 2C6. Lane 1: AIFM2 transfected lysate(40.5 KDa). Lane 2: Non-transfected lysate. Protocol Download Western Blot (Recombinant protein) Protocol Download ELISA Gene Information Entrez GeneID: 84883 GeneBank BC006121 Accession#: Protein AAH06121 Accession#: Gene Name: AIFM2 Gene Alias: AMID,PRG3,RP11-367H5.2 Gene apoptosis-inducing factor, mitochondrion-associated, 2 Description: Omim ID: 605159 Gene Ontology: Hyperlink Gene Summary: The protein encoded by this gene has significant homology to NADH oxidoreductases and the apoptosis-inducing factor PDCD8/AIF. Overexpression of this gene has been shown to induce apoptosis. The expression of this gene is found to be induced by tumor suppressor protein p53 in colon caner cells. -

UNIVERSITY of CALIFORNIA RIVERSIDE Investigations Into The
UNIVERSITY OF CALIFORNIA RIVERSIDE Investigations into the Role of TAF1-mediated Phosphorylation in Gene Regulation A Dissertation submitted in partial satisfaction of the requirements for the degree of Doctor of Philosophy in Cell, Molecular and Developmental Biology by Brian James Gadd December 2012 Dissertation Committee: Dr. Xuan Liu, Chairperson Dr. Frank Sauer Dr. Frances M. Sladek Copyright by Brian James Gadd 2012 The Dissertation of Brian James Gadd is approved Committee Chairperson University of California, Riverside Acknowledgments I am thankful to Dr. Liu for her patience and support over the last eight years. I am deeply indebted to my committee members, Dr. Frank Sauer and Dr. Frances Sladek for the insightful comments on my research and this dissertation. Thanks goes out to CMDB, especially Dr. Bachant, Dr. Springer and Kathy Redd for their support. Thanks to all the members of the Liu lab both past and present. A very special thanks to the members of the Sauer lab, including Silvia, Stephane, David, Matt, Stephen, Ninuo, Toby, Josh, Alice, Alex and Flora. You have made all the years here fly by and made them so enjoyable. From the Sladek lab I want to thank Eugene, John, Linh and Karthi. Special thanks go out to all the friends I’ve made over the years here. Chris, Amber, Stephane and David, thank you so much for feeding me, encouraging me and keeping me sane. Thanks to the brothers for all your encouragement and prayers. To any I haven’t mentioned by name, I promise I haven’t forgotten all you’ve done for me during my graduate years. -

DDEF1 Monoclonal Antibody (M01), Clone 2G7
Produktinformation Diagnostik & molekulare Diagnostik Laborgeräte & Service Zellkultur & Verbrauchsmaterial Forschungsprodukte & Biochemikalien Weitere Information auf den folgenden Seiten! See the following pages for more information! Lieferung & Zahlungsart Lieferung: frei Haus Bestellung auf Rechnung SZABO-SCANDIC Lieferung: € 10,- HandelsgmbH & Co KG Erstbestellung Vorauskassa Quellenstraße 110, A-1100 Wien T. +43(0)1 489 3961-0 Zuschläge F. +43(0)1 489 3961-7 [email protected] • Mindermengenzuschlag www.szabo-scandic.com • Trockeneiszuschlag • Gefahrgutzuschlag linkedin.com/company/szaboscandic • Expressversand facebook.com/szaboscandic DDEF1 monoclonal antibody (M01), clone 2G7 Catalog # : H00050807-M01 規格 : [ 100 ug ] List All Specification Application Image Product Mouse monoclonal antibody raised against a partial recombinant Western Blot (Cell lysate) Description: DDEF1. Immunogen: DDEF1 (NP_060952, 1030 a.a. ~ 1129 a.a) partial recombinant protein with GST tag. MW of the GST tag alone is 26 KDa. Sequence: VQSRDAIQKQASEDSNDLTPTLPETPVPLPRKINTGKNKVRRVKTIYDCQ ADNDDELTFIEGEVIIVTGEEDQEWWIGHIEGQPERKGVFPVSFVHILSD enlarge Western Blot (Cell lysate) Host: Mouse Reactivity: Human, Rat Isotype: IgG2a Kappa Quality Control Antibody Reactive Against Recombinant Protein. enlarge Testing: Western Blot (Recombinant protein) Immunofluorescence enlarge Sandwich ELISA (Recombinant Western Blot detection against Immunogen (36.74 KDa) . protein) Storage Buffer: In 1x PBS, pH 7.4 Storage Store at -20°C or lower. Aliquot to avoid repeated freezing -

Integrated Analysis of the Critical Region 5P15.3–P15.2 Associated with Cri-Du-Chat Syndrome
Genetics and Molecular Biology, 42, 1(suppl), 186-196 (2019) Copyright © 2019, Sociedade Brasileira de Genética. Printed in Brazil DOI: http://dx.doi.org/10.1590/1678-4685-GMB-2018-0173 Research Article Integrated analysis of the critical region 5p15.3–p15.2 associated with cri-du-chat syndrome Thiago Corrêa1, Bruno César Feltes2 and Mariluce Riegel1,3* 1Post-Graduate Program in Genetics and Molecular Biology, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS, Brazil. 2Institute of Informatics, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS, Brazil. 3Medical Genetics Service, Hospital de Clínicas de Porto Alegre, Porto Alegre, RS, Brazil. Abstract Cri-du-chat syndrome (CdCs) is one of the most common contiguous gene syndromes, with an incidence of 1:15,000 to 1:50,000 live births. To better understand the etiology of CdCs at the molecular level, we investigated theprotein–protein interaction (PPI) network within the critical chromosomal region 5p15.3–p15.2 associated with CdCs using systemsbiology. Data were extracted from cytogenomic findings from patients with CdCs. Based on clin- ical findings, molecular characterization of chromosomal rearrangements, and systems biology data, we explored possible genotype–phenotype correlations involving biological processes connected with CdCs candidate genes. We identified biological processes involving genes previously found to be associated with CdCs, such as TERT, SLC6A3, and CTDNND2, as well as novel candidate proteins with potential contributions to CdCs phenotypes, in- cluding CCT5, TPPP, MED10, ADCY2, MTRR, CEP72, NDUFS6, and MRPL36. Although further functional analy- ses of these proteins are required, we identified candidate proteins for the development of new multi-target genetic editing tools to study CdCs. -

Identification of Differentially Expressed Genes in Human Bladder Cancer Through Genome-Wide Gene Expression Profiling
521-531 24/7/06 18:28 Page 521 ONCOLOGY REPORTS 16: 521-531, 2006 521 Identification of differentially expressed genes in human bladder cancer through genome-wide gene expression profiling KAZUMORI KAWAKAMI1,3, HIDEKI ENOKIDA1, TOKUSHI TACHIWADA1, TAKENARI GOTANDA1, KENGO TSUNEYOSHI1, HIROYUKI KUBO1, KENRYU NISHIYAMA1, MASAKI TAKIGUCHI2, MASAYUKI NAKAGAWA1 and NAOHIKO SEKI3 1Department of Urology, Graduate School of Medical and Dental Sciences, Kagoshima University, 8-35-1 Sakuragaoka, Kagoshima 890-8520; Departments of 2Biochemistry and Genetics, and 3Functional Genomics, Graduate School of Medicine, Chiba University, 1-8-1 Inohana, Chuo-ku, Chiba 260-8670, Japan Received February 15, 2006; Accepted April 27, 2006 Abstract. Large-scale gene expression profiling is an effective CKS2 gene not only as a potential biomarker for diagnosing, strategy for understanding the progression of bladder cancer but also for staging human BC. This is the first report (BC). The aim of this study was to identify genes that are demonstrating that CKS2 expression is strongly correlated expressed differently in the course of BC progression and to with the progression of human BC. establish new biomarkers for BC. Specimens from 21 patients with pathologically confirmed superficial (n=10) or Introduction invasive (n=11) BC and 4 normal bladder samples were studied; samples from 14 of the 21 BC samples were subjected Bladder cancer (BC) is among the 5 most common to microarray analysis. The validity of the microarray results malignancies worldwide, and the 2nd most common tumor of was verified by real-time RT-PCR. Of the 136 up-regulated the genitourinary tract and the 2nd most common cause of genes we detected, 21 were present in all 14 BCs examined death in patients with cancer of the urinary tract (1-7). -
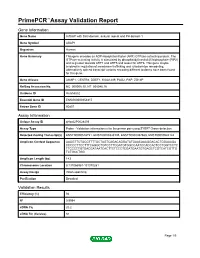
Download Validation Data
PrimePCR™Assay Validation Report Gene Information Gene Name ArfGAP with SH3 domain, ankyrin repeat and PH domain 1 Gene Symbol ASAP1 Organism Human Gene Summary This gene encodes an ADP-ribosylation factor (ARF) GTPase-activating protein. The GTPase-activating activity is stimulated by phosphatidylinositol 45-biphosphate (PIP2) and is greater towards ARF1 and ARF5 and lesser for ARF6. This gene maybe involved in regulation of membrane trafficking and cytoskeleton remodeling. Alternatively spliced transcript variants encoding different isoforms have been found for this gene. Gene Aliases AMAP1, CENTB4, DDEF1, KIAA1249, PAG2, PAP, ZG14P RefSeq Accession No. NC_000008.10, NT_008046.16 UniGene ID Hs.655552 Ensembl Gene ID ENSG00000153317 Entrez Gene ID 50807 Assay Information Unique Assay ID qHsaCIP0028499 Assay Type Probe - Validation information is for the primer pair using SYBR® Green detection Detected Coding Transcript(s) ENST00000518721, ENST00000343135, ENST00000357668, ENST00000524124 Amplicon Context Sequence AAGGTTCTGCGTTTTGCTAGTCAGACAGGATATGAACAAAGGACACTGGAAAGA CCCCCTTCCTTTCAGGCTGTCCTTCGATGTGGCCAATCCACCACTCCTGGTCCTC TTCCCCTGTGACGATAATCACTTCTCCCTCGATGAATGTGAGCTCGTCATCGTTG TCTGCCTGG Amplicon Length (bp) 143 Chromosome Location 8:131066961-131070281 Assay Design Intron-spanning Purification Desalted Validation Results Efficiency (%) 94 R2 0.9994 cDNA Cq 20.2 cDNA Tm (Celsius) 84 Page 1/5 PrimePCR™Assay Validation Report gDNA Cq 38.05 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. -

Comparative Analyses of Gene Copy Number and Mrna Expression in GBM Tumors And
Title page Comparative analyses of gene copy number and mRNA expression in GBM tumors and GBM xenografts J. Graeme Hodgson # *, Ru-Fang Yeh #, Amrita Ray, Nicholas J Wang, Ivan Smirnov, Mamie Yu, Sujatmi Hariono, Joachim Silber, Heidi S. Feiler, Joe W. Gray, Paul T. Spellman, Scott R. Vandenberg, Mitchel S. Berger, C. David James Departments of Neurological Surgery (JGH, IS, MY, SH, JS, SRV, MSB, CDJ), Pathology (SRV), and Epidemiology and Biostatistics (RY) University of California, San Francisco, CA 94143. Life Sciences Division, Lawrence Berkeley National Lab, Berkeley, CA 94720 (AR, NJW, HSF, JWG, PTS) # These authors contributed equally to this work Running title: Genomic analyses of a GBM xenograft tumor panel * Corresponding Author J. Graeme Hodgson Dept. Neurological Surgery UC San Francisco San Francisco, CA 94143-0808 Phone: 415-476-3630 Fax: 415-476-8218 e-mail: [email protected] Abstract Development of model systems that recapitulate the molecular heterogeneity observed amongst GBM tumors will expedite the testing of targeted molecular therapeutic strategies for GBM treatment. In this study, we profiled DNA copy number and mRNA expression in 21 independent GBM tumor lines maintained as subcutaneous xenografts (GBMX), and compared GBMX molecular signatures to those observed in GBM clinical specimens derived from The Cancer Genome Atlas (TCGA). The predominant copy number signature in both tumor groups was defined by chromosome-7-gain/chromosome-10-loss, a poor prognosis genetic signature. We also observed, at frequencies similar to that detected in TCGA GBMs genomic amplification and overexpression of known GBM oncogenes such as EGFR, MDM2, CDK6 and MYCN, and novel genes including NUP107, SLC35E3, MMP1, MMP13 and DDX1. -

A Dissertation Submitted to the Faculty of the Graduate School of Arts And
MOLECULAR MODULATION OF ESTROGEN -INDUCED APOPTOSIS IN LONG -TERM ESTROGEN - DEPRIVED BREAST CANCER CELLS A Dissertation submitted to the Faculty of the Graduate School of Arts and Sciences of Georgetown University in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Tumor Biology By Elizabeth E. Sweeney, B.S. Washington, DC March 31, 2014 Copyright 2014 by Elizabeth E. Sweeney All Rights Reserved ii MOLECULAR MODULATION OF ESTROGEN -INDUCED APOPTOSIS IN LONG -TERM ESTROGEN - DEPRIVED BREAST CANCER CELLS Elizabeth E. Sweeney, B.S. Thesis Advisor: V. Craig Jordan , O.B.E., Ph.D., D.Sc., F.Med.Sci . ABSTRACT Estrogen receptor (ER)-positive breast cancer cell lines have been instrumental in modeling breast cancer and providing an opportunity to document the development and evolution of acquired anti-hormone resistance. Models of long-term estrogen-deprived breast cancer cells are utilized in the laboratory to mimic clinical aromatase inhibitor-resistant breast cancer and serve as a tool to discover new therapeutic strategies. The MCF-7:5C and MCF-7:2A subclones were generated through long-term estrogen deprivation of ER-positive MCF-7 cells, and represent anti-hormone resistant breast cancer. MCF-7:5C cells paradoxically undergo estrogen-induced apoptosis within seven days of estrogen (estradiol, E 2) treatment; MCF-7:2A cells also experience E2-induced apoptosis but evade dramatic cell death until approximately 14 days of treatment. Our data suggest that MCF-7:2A cells employ stronger antioxidant defense mechanisms than do MCF-7:5C cells, and that oxidative stress is ultimately required for MCF- 7:2A cells to die in response to E2 treatment. -

Ferroptosis-Related Flavoproteins: Their Function and Stability
International Journal of Molecular Sciences Review Ferroptosis-Related Flavoproteins: Their Function and Stability R. Martin Vabulas Charité-Universitätsmedizin, Institute of Biochemistry, Charitéplatz 1, 10117 Berlin, Germany; [email protected]; Tel.: +49-30-4505-28176 Abstract: Ferroptosis has been described recently as an iron-dependent cell death driven by peroxida- tion of membrane lipids. It is involved in the pathogenesis of a number of diverse diseases. From the other side, the induction of ferroptosis can be used to kill tumor cells as a novel therapeutic approach. Because of the broad clinical relevance, a comprehensive understanding of the ferroptosis-controlling protein network is necessary. Noteworthy, several proteins from this network are flavoenzymes. This review is an attempt to present the ferroptosis-related flavoproteins in light of their involvement in anti-ferroptotic and pro-ferroptotic roles. When available, the data on the structural stability of mutants and cofactor-free apoenzymes are discussed. The stability of the flavoproteins could be an important component of the cellular death processes. Keywords: flavoproteins; riboflavin; ferroptosis; lipid peroxidation; protein quality control 1. Introduction Human flavoproteome encompasses slightly more than one hundred enzymes that par- ticipate in a number of key metabolic pathways. The chemical versatility of flavoproteins relies on the associated cofactors, flavin mononucleotide (FMN) and flavin adenine dinu- cleotide (FAD). In humans, flavin cofactors are biosynthesized from a precursor riboflavin that has to be supplied with food. To underline its nutritional essentiality, riboflavin is called vitamin B2. In compliance with manifold cellular demands, flavoproteins have been accommo- Citation: Vabulas, R.M. dated to operate at different subcellular locations [1].