Microbial Reductive Transformation Of
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Exposure and Use Assessment for Five PBT Chemicals
EPA Document # EPA-740-R1-8002 June 2018 United States Office of Chemical Safety and Environmental Protection Agency Pollution Prevention Exposure and Use Assessment of Five Persistent, Bioaccumulative and Toxic Chemicals Peer Review Draft June 2018 Contents TABLES ................................................................................................................................................................... 7 FIGURES ................................................................................................................................................................. 7 1. EXECUTIVE SUMMARY ................................................................................................................................ 15 2. BACKGROUND ............................................................................................................................................. 15 3. APPROACH .................................................................................................................................................. 17 4. DECABROMODIPHENYL ETHER (DECABDE) .................................................................................................. 21 4.1. Chemistry and Physical-Chemical Properties ................................................................................ 21 4.2. Uses ................................................................................................................................................ 21 4.3. Characterization of Expected Environmental Partitioning -

List of Lists
United States Office of Solid Waste EPA 550-B-10-001 Environmental Protection and Emergency Response May 2010 Agency www.epa.gov/emergencies LIST OF LISTS Consolidated List of Chemicals Subject to the Emergency Planning and Community Right- To-Know Act (EPCRA), Comprehensive Environmental Response, Compensation and Liability Act (CERCLA) and Section 112(r) of the Clean Air Act • EPCRA Section 302 Extremely Hazardous Substances • CERCLA Hazardous Substances • EPCRA Section 313 Toxic Chemicals • CAA 112(r) Regulated Chemicals For Accidental Release Prevention Office of Emergency Management This page intentionally left blank. TABLE OF CONTENTS Page Introduction................................................................................................................................................ i List of Lists – Conslidated List of Chemicals (by CAS #) Subject to the Emergency Planning and Community Right-to-Know Act (EPCRA), Comprehensive Environmental Response, Compensation and Liability Act (CERCLA) and Section 112(r) of the Clean Air Act ................................................. 1 Appendix A: Alphabetical Listing of Consolidated List ..................................................................... A-1 Appendix B: Radionuclides Listed Under CERCLA .......................................................................... B-1 Appendix C: RCRA Waste Streams and Unlisted Hazardous Wastes................................................ C-1 This page intentionally left blank. LIST OF LISTS Consolidated List of Chemicals -

Reporting Requirements for Hazardous Substances and List of Hazardous Substances
1301:7-9-03 Reporting requirements for hazardous substances and list of hazardous substances. (A) Purpose. For the purpose of prescribing rules pursuant to section 3737.88 of the Revised Code, the fire marshal hereby adopts this rule to establish reporting requirements for underground storage tank systems that contain hazardous substance(s) and to list those substances which are hereby identified as hazardous substances. This rule is adopted by the fire marshal in accordance with Chapter 119 of the Revised Code and shall not be considered a part of the "Ohio Fire Code". (B) Definitions. For the purpose of this rule: (1) "Release of a hazardous substance" means: (a) Any spilling, leaking, emitting, discharging, escaping, leaching or disposing of a hazardous substance(s) from an underground storage tank system into the ground water, a surface water body, subsurface soils or otherwise into the environment; (b) Any spilling, leaking, emitting, discharging, escaping, or disposing of a hazardous substance(s) into ground water, a surface water body, subsurface soils or otherwise into the environment while transferring or attempting to transfer a hazardous substance(s) into an underground storage tank system; or (c) Contamination of subsurface soils or ground water on the UST site by a hazardous substance(s) found and confirmed through laboratory analysis of samples from the UST site. (2) "Suspected release of a hazardous substance" means evidence of a release of a hazardous substance(s) obtained through one or more of the following events: (a) -

List of Hazardous Substances and Reportable Quantities 1. This Appendix Lists Materials
172.101 App A Appendix A to §172.101 - List of Hazardous Substances and Reportable Quantities 1. This Appendix lists materials and their corresponding reportable quantities (RQ's) that are listed or designated as "hazardous substances'' under section 101(14) of the Comprehensive Environmental Response, Compensation, and Liability Act, 42 U.S.C. 9601(14) (CERCLA; 42 U.S.C. 9601 et seq). This listing fulfills the requirement of CERCLA, 42 U.S.C. 9656(a), that all ``hazardous substances,'' as defined in 42 U.S.C. 9601(14), be listed and regulated as hazardous materials under 49 U.S.C. 5101-5127. That definition includes substances listed under sections 311(b)(2)(A) and 307(a) of the Federal Water Pollution Control Act, 33 U.S.C. 1321(b)(2)(A) and 1317(a), section 3001 of the Solid Waste Disposal Act, 42 U.S.C. 6921, and section 112 of the Clean Air Act, 42 U.S.C. 7412. In addition, this list contains materials that the Administrator of the Environmental Protection Agency has determined to be hazardous substances in accordance with section 102 of CERCLA, 42 U.S.C. 9602. It should be noted that 42 U.S.C. 9656(b) provides that common and contract carriers may be held liable under laws other than CERCLA for the release of a hazardous substance as defined in that Act, during transportation that commenced before the effective date of the listing and regulating of that substance as a hazardous material under 49 U.S.C. 5101-5127. 2. This Appendix is divided into two TABLES which are entitled "TABLE 1-HAZARDOUS SUBSTANCES OTHER THAN RADIONUCLIDES'' and "TABLE 2-RADIONUCLIDES.'' A material listed in this Appendix is regulated as a hazardous material and a hazardous substance under this subchapter if it meets the definition of a hazardous substance in §171.8 of this subchapter. -

Sheet Materials Chemical Compatibility Chart
Sheet Materials Chemical Compatibility Chart Revised to match Compressed Fiber & Graphite Sheet Brochure (US), and to reflect any possible attack on material binder (NBR, inc THERMICULITE). Now includes SF4300. Key: A = Suitable As a general guide, TH 835 Spirals and TH845 Flexpros may be assumed B = Depends on operating conditions** to have the same compatibility as TH 815; however, any variations B*= Corrosion of SS316 core possible under certain conditions in winding metal or core from standard SS316 must be taken into account C = Not suitable ** One or more components of material may be degraded. Higher temperature will exacerbate attack; higher concentration may exacerbate attack if viscosity of medium does not increase significantly with concentration; higher gasket load will mitigate attack (limited to edge in contact with media) The given ratings apply only to materials adequately clamped between flanges, and not to freely - immersed samples SIGMA® Thermiculite Compressed Fiber SF2401 SF3300 Flexicarb SF3500 Medium 500/501 511 533 588 600 815 715 (FG) SF4300 SF2420 SF5000 TH714 AF2100 Abietic Acid A A A A A A A A A A A A A Acetaldehyde A A A A A B B A B B B A B Acetamide A A A A A B B A B B B A B Acetic Acid (Crude, Glacial, Pure) A A A A A B B A B B B A B Acetic Anhydride A A A A A B B A B B B A B Acetone A A A A A B B A B B B A B Acetonitrile A A A A A B B A B B B A B Acetophenone A A A A A B B A B B B A B 2-Acetylaminofluorene A A A A A B B A B B B A B Acetylene A A A A A A A A A B A A A Acrolein A A A A A B B A B B B A B Acrylamide -

Chemical Specific Parameters May 2020
Regional Screening Level (RSL) Chemical-specific Parameters Supporting Table May 2020 Contaminant Molecular Weight Volatility Parameters Melting Point Density Diffusivity in Air and Water Partition Coefficients Water Solubility Tapwater Dermal Parameters MW H` HLC H`and HLC VP VP MP MP Density Density Dia Diw Dia and Diw Kd Kd Koc Koc log Kow log Kow S S B τevent t* Kp Kp Analyte CAS No. MW Ref (unitless) (atm-m3/mole) Ref mmHg Ref C Ref (g/cm3) Ref (cm2/s) (cm2/s) Ref (L/kg) Ref (L/kg) Ref (unitless) Ref (mg/L) Ref (unitless) (hr/event) (hr) (cm/hr) Ref Acephate 30560-19-1 1.8E+02 PHYSPROP 2.0E-11 5.0E-13 EPI 1.7E-06 PHYSPROP 8.8E+01 PHYSPROP 1.4E+00 CRC89 3.7E-02 8.0E-06 WATER 9 1.0E+01 EPI -8.5E-01 PHYSPROP 8.2E+05 PHYSPROP 2.1E-04 1.1E+00 2.7E+00 4.0E-05 EPI Acetaldehyde 75-07-0 4.4E+01 PHYSPROP 2.7E-03 6.7E-05 PHYSPROP 9.0E+02 PHYSPROP -1.2E+02 PHYSPROP 7.8E-01 CRC89 1.3E-01 1.4E-05 WATER 9 1.0E+00 EPI -3.4E-01 PHYSPROP 1.0E+06 PHYSPROP 1.3E-03 1.9E-01 4.5E-01 5.3E-04 EPI Acetochlor 34256-82-1 2.7E+02 PHYSPROP 9.1E-07 2.2E-08 PHYSPROP 2.8E-05 PHYSPROP 1.1E+01 PubChem 1.1E+00 PubChem 2.2E-02 5.6E-06 WATER 9 3.0E+02 EPI 3.0E+00 PHYSPROP 2.2E+02 PHYSPROP 3.1E-02 3.4E+00 8.2E+00 5.0E-03 EPI Acetone 67-64-1 5.8E+01 PHYSPROP 1.4E-03 3.5E-05 PHYSPROP 2.3E+02 PHYSPROP -9.5E+01 PHYSPROP 7.8E-01 CRC89 1.1E-01 1.1E-05 WATER 9 2.4E+00 EPI -2.4E-01 PHYSPROP 1.0E+06 PHYSPROP 1.5E-03 2.2E-01 5.3E-01 5.1E-04 EPI Acetone Cyanohydrin 75-86-5 8.5E+01 PHYSPROP 8.1E-08 2.0E-09 PHYSPROP 3.4E-01 PHYSPROP -1.9E+01 PHYSPROP 9.3E-01 CRC89 8.6E-02 1.0E-05 -
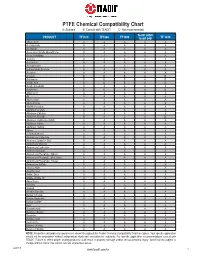
Chemical Compatibility Chart
PTFE Chemical Compatibility Chart A: Suitable B: Consult with TEADIT C: Not recommended Teadit 24SH PRODUCT TF1570 TF1580 TF1590 Teadit 24B TF 1510 Abietic Acid A A A A A Acetaldehyde A A A A A Acetamide A A A A A Acetic Acid (Crude,Glacial,Pure) A A A A A Acetic Anhydride A A A A A Acetone A A A A A Acetonitrile A A A A A Acetophenone A A A A A 2-Acetylaminofluorene A A A A A Acetylene A A A A A Acrolein B B B A B Acrylamide B B B A B Acrylic Acid B B B A B Acrylic Anhydride A A A A A Acrylonitrile B B B A B Adipic Acid A A A A A Air A A A A A Allyl Acetate A A A A A Allyl Chloride A A A A A Allyl Methacrylate A A A A A Aluminum Acetate A A A A A Aluminum Chloride A A A A A Aluminum Fluoride B A C A B Aluminum Hydroxide (Solid) A A A A A Aluminum Nitrate A A A A A Aluminum Sulfate A A A A A Alums A A A A A 4-Aminodiphenyl A A A A A Ammonium Carbonate A A A A A Ammonia, Liquid or Gas A A A A A Ammonium Chloride A A A A A Ammonium Hydroxide A A A A A Ammonium Nitrate A A A A A Ammonium Phosphate, Dibasic A A A A A Ammonium Phosphate, Monobasic A A A A A Ammonium Phosphate, Tribasic A A A A A Ammonium Sulfate A A A A A Amyl Acetate A A A A A Amyl Alcohol A A A A A Aniline Dyes A A A A A Aniline, Aniline Oil A A A A A Aqua Regia A A A A A Aroclors A A A A A Asphalt A A A A A Aviation Gasoline A A A A A Barium Chloride A A A A A Barium Hydroxide A A A A A Barium Sulfide A A A A A Beer A A A A A Benzaldehyde A A A A A Benzene, Benzol A A A A A Benzidine A A A A A Benzoic Acid A A A A A Benzonitrile A A A A A Benzotrichloride A A A A A Benzoyl Chloride A A A A A NOTE: Properties and application parameters shown throughout this Tealon Chemical Compatibility Chart are typical. -

6 NYCRR Part
6 NYCRR PART 597 HAZARDOUS SUBSTANCES IDENTIFICATION, RELEASE PROHIBITION, AND RELEASE REPORTING (Statutory authority: Environmental Conservation Law sections 1-0101, 3-0301, 3-0303, 17-0301, 17-0303, 17-0501, 17-1743, 37-0101 through 37-0107, and 40-0101 through 40-0121) TABLE OF CONTENTS 597.1 General. ....................................................................................................................2 597.2 Criteria for identifying a hazardous substance or acutely hazardous substance. .....5 597.3 List of hazardous substances. ...................................................................................6 597.4 Spills and Releases of hazardous substances. ..........................................................6 Table 1 Hazardous Substances – Sorted by Name ................................................................9 Table 2 Hazardous Substances – Sorted by CAS Number .................................................49 6 NYCRR Part 597 – Oct. 11, 2015 Page 1 of 88 597.1 597.1 General. (a) Purpose. The purpose of this Part is to: (1) set forth criteria for identifying a hazardous substance or acutely hazardous substance; (2) set forth a list of hazardous substances; (3) identify reportable quantities for the spill or release of hazardous substances; (4) prohibit the unauthorized release of hazardous substances; and (5) establish requirements for reporting of releases of hazardous substances. (b) Definitions. The following definitions apply to this Part: (1) Authorized means the possession of a valid license, permit, or certificate issued by an agency of the state of New York or the federal government, or an order issued by the Department or United States Environmental Protection Agency under applicable statutes, rules or regulations regarding the possession or release of hazardous substances or otherwise engaging in conduct which is exempt under applicable statutes, rules or regulations from the requirements of possessing such a license, permit, certificate or order. -

Epa Hazardous Waste Codes
EPA HAZARDOUS WASTE CODES Code Waste description Code Waste description D001 Ignitable waste D023 o-Cresol D002 Corrosive waste D024 m-Cresol D003 Reactive waste D025 p-Cresol D004 Arsenic D026 Cresol D005 Barium D027 1,4-Dichlorobenzene D006 Cadmium D028 1,2-Dichloroethane D007 Chromium D029 1,1-Dichloroethylene D008 Lead D030 2,4-Dinitrotoluene D009 Mercury D031 Heptachlor (and its epoxide) D010 Selenium D032 Hexachlorobenzene D011 Silver D033 Hexachlorobutadiene D012 Endrin(1,2,3,4,10,10-hexachloro-1,7-epoxy- D034 Hexachloroethane 1,4,4a,5,6,7,8,8a-octahydro-1,4-endo, endo- 5,8-dimeth-ano-naphthalene) D035 Methyl ethyl ketone D013 Lindane (1,2,3,4,5,6-hexa- D036 Nitrobenzene chlorocyclohexane, gamma isomer) D037 Pentachlorophenol D014 Methoxychlor (1,1,1-trichloro-2,2-bis [p- methoxyphenyl] ethane) D038 Pyridine D015 Toxaphene (C10 H10 Cl8, Technical D039 Tetrachloroethylene chlorinated camphene, 67-69 percent chlorine) D040 Trichlorethylene D016 2,4-D (2,4-Dichlorophenoxyacetic acid) D041 2,4,5-Trichlorophenol D017 2,4,5-TP Silvex (2,4,5- D042 2,4,6-Trichlorophenol Trichlorophenoxypropionic acid) D043 Vinyl chloride D018 Benzene D019 Carbon tetrachloride D020 Chlordane D021 Chlorobenzene D022 Chloroform B-1 EPA HAZARDOUS WASTE CODES (Continued) Code Waste description Code Waste description HAZARDOUS WASTE FROM NONSPECIFIC solvents: cresols, cresylic acid, and SOURCES nitrobenzene; and the still bottoms from the recovery of these solvents; all spent solvent F001 The following spent halogenated solvents mixtures/blends containing, before use, a used in degreasing: Tetrachloroethylene, total of ten percent or more (by volume) of trichlorethylene, methylene chloride, 1,1,1- one or more of the above nonhalogenated trichloroethane, carbon tetrachloride and solvents or those solvents listed in F001, chlorinated fluorocarbons; all spent solvent F002, and F005; and still bottoms from the mixtures/blends used in degreasing recovery of these spent solvents and spent containing, before use, a total of ten solvent mixtures. -
Hybrid Computational Toxicology Models for Regulatory Risk Assessment Prachi Pradeep Marquette University
Marquette University e-Publications@Marquette Dissertations (2009 -) Dissertations, Theses, and Professional Projects Hybrid Computational Toxicology Models for Regulatory Risk Assessment Prachi Pradeep Marquette University Recommended Citation Pradeep, Prachi, "Hybrid Computational Toxicology Models for Regulatory Risk Assessment" (2015). Dissertations (2009 -). Paper 503. http://epublications.marquette.edu/dissertations_mu/503 HYBRID COMPUTATIONAL TOXICOLOGY MODELS FOR REGULATORY RISK ASSESSMENT by PRACHI PRADEEP A Dissertation submitted to the Faculty of the Graduate School, Marquette University, in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy Milwaukee, Wisconsin May 2015 ABSTRACT HYBRID COMPUTATIONAL TOXICOLOGY MODELS FOR REGULATORY RISK ASSESSMENT Prachi Pradeep Marquette University, 2015 Computational toxicology is the development of quantitative structure activity relationship (QSAR) models that relate a quantitative measure of chemical structure to a biological effect. In silico QSAR tools are widely accepted as a faster alternative to time-consuming clinical and animal testing methods for regulatory risk assessment of xenobiotics used in consumer products. However, different QSAR tools often make contrasting predictions for a new xenobiotic and may also vary in their predictive ability for different class of xenobiotics. This makes their use challenging, especially in regulatory applications, where transparency and interpretation of predictions play a crucial role in the development of safety -

UNITED NATIONS Rotterdam Convention on the Prior Informed
UNITED NATIONS RC UNEP/FAO/RC/CRC.7/9/Add.1 Rotterdam Convention on the Prior Distr.: General 20 December 2010 Informed Consent Procedure for Certain Hazardous Chemicals and English only Pesticides in International Trade Chemical Review Committee Seventh meeting Rome, 28 March–1 April 2011 Item 4 (c) (vi) of the provisional agenda∗ Technical work: review of notifications of final regulatory action: pentachlorobenzene Pentachlorobenzene Note by the Secretariat Addendum Supporting documentation provided by Canada The Secretariat has the honour to provide, in the annex to the present note, documentation received from Canada to support its notification of final regulatory action for pentachlorobenzene as an industrial chemical. The documentation has not been formally edited. ∗ UNEP/FAO/RC/CRC.7/1. K1063386 280111 UNEP/FAO/RC/CRC.7/9/Add.1 Annex ____________________ 2 ChemBioFinder.Com- - Structure Details Page 1 of 1 CambridgeSoft.com ChemBioNews.com ChemBioFinder.com SciStore.com Login Register Reference Chemicals Reactions Software About Help Contact Home Record 1 of 1 New Query Return to List Molecular Profile Report Resources View Record Content Type Source Database Key Subscribe Reference ChemIndex (CI) 608-93-5 Subscribe Reference National Cancer Institute (NCI) 1857 View Record Reference NetProtm Molecular Connections (NETPRO) MC50882 Subscribe Safety Sigma Aldrich MSDS (SAMSDS) X1003313-5 Subscribe Sourcing ChemACX (ACX) X1003313-5 Names and Synonyms Names Sources 1,2,3,4,5-Pentachlorobenzene CHEMACX CHEMINDEX NCI NETPRO Benzene, pentachloro- (8CI9CI) NCI Pentachlorobenzene CHEMACX CHEMINDEX NCI SA-MSDS QCB CHEMACX CHEMINDEX NCI WLN: GR BG CG DG EG NCI Identifiers Chemical Identifiers Type Sources X1003313-5 acx_id SA-MSDS CHEMACX 608-93-5 casrn CHEMACX CHEMINDEX NETPRO NCI SA-MSDS MC50882 int_id NETPRO 608-93-5 mol_rel_id CHEMACX CHEMINDEX NETPRO NCI SA-MSDS 1857 nsc NCI Properites Type Value Source Formula C6HCl5 CSCARTRIDGE MW 250.3371 CSCARTRIDGE SMILES ClC1=CC(Cl)=C(Cl)C(Cl)=C1Cl CSCARTRIDGE Std. -

Toxicological Profile for Pentachlorophenol; Thus, the Literature Search Was Restricted to Studies Published Between January 1999 and January 2019
Toxicological Profile for Pentachlorophenol Draft for Public Comment July 2021 PENTACHLOROPHENOL ii DISCLAIMER Use of trade names is for identification only and does not imply endorsement by the Agency for Toxic Substances and Disease Registry, the Public Health Service, or the U.S. Department of Health and Human Services. This information is distributed solely for the purpose of pre dissemination public comment under applicable information quality guidelines. It has not been formally disseminated by the Agency for Toxic Substances and Disease Registry. It does not represent and should not be construed to represent any agency determination or policy. ***DRAFT FOR PUBLIC COMMENT*** PENTACHLOROPHENOL iii FOREWORD This toxicological profile is prepared in accordance with guidelines developed by the Agency for Toxic Substances and Disease Registry (ATSDR) and the Environmental Protection Agency (EPA). The original guidelines were published in the Federal Register on April 17, 1987. Each profile will be revised and republished as necessary. The ATSDR toxicological profile succinctly characterizes the toxicologic and adverse health effects information for these toxic substances described therein. Each peer-reviewed profile identifies and reviews the key literature that describes a substance's toxicologic properties. Other pertinent literature is also presented, but is described in less detail than the key studies. The profile is not intended to be an exhaustive document; however, more comprehensive sources of specialty information are referenced. The focus of the profiles is on health and toxicologic information; therefore, each toxicological profile begins with a relevance to public health discussion which would allow a public health professional to make a real-time determination of whether the presence of a particular substance in the environment poses a potential threat to human health.