Novel Prognostic Biomarkers of Gastric Cancer Based on Gene Expression Microarray: COL12A1, GSTA3, FGA and FGG
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Chuanxiong Rhizoma Compound on HIF-VEGF Pathway and Cerebral Ischemia-Reperfusion Injury’S Biological Network Based on Systematic Pharmacology
ORIGINAL RESEARCH published: 25 June 2021 doi: 10.3389/fphar.2021.601846 Exploring the Regulatory Mechanism of Hedysarum Multijugum Maxim.-Chuanxiong Rhizoma Compound on HIF-VEGF Pathway and Cerebral Ischemia-Reperfusion Injury’s Biological Network Based on Systematic Pharmacology Kailin Yang 1†, Liuting Zeng 1†, Anqi Ge 2†, Yi Chen 1†, Shanshan Wang 1†, Xiaofei Zhu 1,3† and Jinwen Ge 1,4* Edited by: 1 Takashi Sato, Key Laboratory of Hunan Province for Integrated Traditional Chinese and Western Medicine on Prevention and Treatment of 2 Tokyo University of Pharmacy and Life Cardio-Cerebral Diseases, Hunan University of Chinese Medicine, Changsha, China, Galactophore Department, The First 3 Sciences, Japan Hospital of Hunan University of Chinese Medicine, Changsha, China, School of Graduate, Central South University, Changsha, China, 4Shaoyang University, Shaoyang, China Reviewed by: Hui Zhao, Capital Medical University, China Background: Clinical research found that Hedysarum Multijugum Maxim.-Chuanxiong Maria Luisa Del Moral, fi University of Jaén, Spain Rhizoma Compound (HCC) has de nite curative effect on cerebral ischemic diseases, *Correspondence: such as ischemic stroke and cerebral ischemia-reperfusion injury (CIR). However, its Jinwen Ge mechanism for treating cerebral ischemia is still not fully explained. [email protected] †These authors share first authorship Methods: The traditional Chinese medicine related database were utilized to obtain the components of HCC. The Pharmmapper were used to predict HCC’s potential targets. Specialty section: The CIR genes were obtained from Genecards and OMIM and the protein-protein This article was submitted to interaction (PPI) data of HCC’s targets and IS genes were obtained from String Ethnopharmacology, a section of the journal database. -

Inhibition of Mitochondrial Complex II in Neuronal Cells Triggers Unique
www.nature.com/scientificreports OPEN Inhibition of mitochondrial complex II in neuronal cells triggers unique pathways culminating in autophagy with implications for neurodegeneration Sathyanarayanan Ranganayaki1, Neema Jamshidi2, Mohamad Aiyaz3, Santhosh‑Kumar Rashmi4, Narayanappa Gayathri4, Pulleri Kandi Harsha5, Balasundaram Padmanabhan6 & Muchukunte Mukunda Srinivas Bharath7* Mitochondrial dysfunction and neurodegeneration underlie movement disorders such as Parkinson’s disease, Huntington’s disease and Manganism among others. As a corollary, inhibition of mitochondrial complex I (CI) and complex II (CII) by toxins 1‑methyl‑4‑phenylpyridinium (MPP+) and 3‑nitropropionic acid (3‑NPA) respectively, induced degenerative changes noted in such neurodegenerative diseases. We aimed to unravel the down‑stream pathways associated with CII inhibition and compared with CI inhibition and the Manganese (Mn) neurotoxicity. Genome‑wide transcriptomics of N27 neuronal cells exposed to 3‑NPA, compared with MPP+ and Mn revealed varied transcriptomic profle. Along with mitochondrial and synaptic pathways, Autophagy was the predominant pathway diferentially regulated in the 3‑NPA model with implications for neuronal survival. This pathway was unique to 3‑NPA, as substantiated by in silico modelling of the three toxins. Morphological and biochemical validation of autophagy markers in the cell model of 3‑NPA revealed incomplete autophagy mediated by mechanistic Target of Rapamycin Complex 2 (mTORC2) pathway. Interestingly, Brain Derived Neurotrophic Factor -

GSTA3 (NM 000847) Human Tagged ORF Clone Lentiviral Particle Product Data
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for RC204624L4V GSTA3 (NM_000847) Human Tagged ORF Clone Lentiviral Particle Product data: Product Type: Lentiviral Particles Product Name: GSTA3 (NM_000847) Human Tagged ORF Clone Lentiviral Particle Symbol: GSTA3 Synonyms: GSTA3-3; GTA3 Vector: pLenti-C-mGFP-P2A-Puro (PS100093) ACCN: NM_000847 ORF Size: 666 bp ORF Nucleotide The ORF insert of this clone is exactly the same as(RC204624). Sequence: OTI Disclaimer: The molecular sequence of this clone aligns with the gene accession number as a point of reference only. However, individual transcript sequences of the same gene can differ through naturally occurring variations (e.g. polymorphisms), each with its own valid existence. This clone is substantially in agreement with the reference, but a complete review of all prevailing variants is recommended prior to use. More info OTI Annotation: This clone was engineered to express the complete ORF with an expression tag. Expression varies depending on the nature of the gene. RefSeq: NM_000847.3 RefSeq Size: 915 bp RefSeq ORF: 669 bp Locus ID: 2940 UniProt ID: Q16772 Protein Pathways: Drug metabolism - cytochrome P450, Glutathione metabolism, Metabolism of xenobiotics by cytochrome P450 MW: 25.3 kDa This product is to be used for laboratory only. Not for diagnostic or therapeutic use. View online » ©2021 OriGene Technologies, Inc., 9620 Medical Center Drive, Ste 200, Rockville, MD 20850, US 1 / 2 GSTA3 (NM_000847) Human Tagged ORF Clone Lentiviral Particle – RC204624L4V Gene Summary: Cytosolic and membrane-bound forms of glutathione S-transferase are encoded by two distinct supergene families. -

Comparative Response of the Hepatic Transcriptomes of Domesticated and Wild Turkey to Aflatoxin B1
toxins Article Comparative Response of the Hepatic Transcriptomes of Domesticated and Wild Turkey to Aflatoxin B1 Kent M. Reed 1,* ID , Kristelle M. Mendoza 1, Juan E. Abrahante 2 and Roger A. Coulombe 3 1 Department of Veterinary and Biomedical Sciences, College of Veterinary Medicine, University of Minnesota, Saint Paul, MN 55108, USA; [email protected] 2 University of Minnesota Informatics Institute, University of Minnesota, Minneapolis, MN 55455, USA; [email protected] 3 Department of Animal, Dairy and Veterinary Sciences, College of Agriculture, Utah State University, Logan, UT 84322, USA; [email protected] * Correspondence: [email protected]; Tel.: +1-612-624-1287; Fax: +1-612-625-0204 Received: 29 November 2017; Accepted: 10 January 2018; Published: 13 January 2018 Abstract: The food-borne mycotoxin aflatoxin B1 (AFB1) poses a significant risk to poultry, which are highly susceptible to its hepatotoxic effects. Domesticated turkeys (Meleagris gallopavo) are especially sensitive, whereas wild turkeys (M. g. silvestris) are more resistant. AFB1 toxicity entails bioactivation by hepatic cytochrome P450s to the electrophilic exo-AFB1-8,9-epoxide (AFBO). Domesticated turkeys lack functional hepatic GST-mediated detoxification of AFBO, and this is largely responsible for the differences in resistance between turkey types. This study was designed to characterize transcriptional changes induced in turkey livers by AFB1, and to contrast the response of domesticated (susceptible) and wild (more resistant) birds. Gene expression responses to AFB1 were examined using RNA-sequencing. Statistically significant differences in gene expression were observed among treatment groups and between turkey types. Expression analysis identified 4621 genes with significant differential expression (DE) in AFB1-treated birds compared to controls. -

Comprehensive Analysis Reveals Novel Gene Signature in Head and Neck Squamous Cell Carcinoma: Predicting Is Associated with Poor Prognosis in Patients
5892 Original Article Comprehensive analysis reveals novel gene signature in head and neck squamous cell carcinoma: predicting is associated with poor prognosis in patients Yixin Sun1,2#, Quan Zhang1,2#, Lanlin Yao2#, Shuai Wang3, Zhiming Zhang1,2 1Department of Breast Surgery, The First Affiliated Hospital of Xiamen University, School of Medicine, Xiamen University, Xiamen, China; 2School of Medicine, Xiamen University, Xiamen, China; 3State Key Laboratory of Cellular Stress Biology, School of Life Sciences, Xiamen University, Xiamen, China Contributions: (I) Conception and design: Y Sun, Q Zhang; (II) Administrative support: Z Zhang; (III) Provision of study materials or patients: Y Sun, Q Zhang; (IV) Collection and assembly of data: Y Sun, L Yao; (V) Data analysis and interpretation: Y Sun, S Wang; (VI) Manuscript writing: All authors; (VII) Final approval of manuscript: All authors. #These authors contributed equally to this work. Correspondence to: Zhiming Zhang. Department of Surgery, The First Affiliated Hospital of Xiamen University, Xiamen, China. Email: [email protected]. Background: Head and neck squamous cell carcinoma (HNSC) remains an important public health problem, with classic risk factors being smoking and excessive alcohol consumption and usually has a poor prognosis. Therefore, it is important to explore the underlying mechanisms of tumorigenesis and screen the genes and pathways identified from such studies and their role in pathogenesis. The purpose of this study was to identify genes or signal pathways associated with the development of HNSC. Methods: In this study, we downloaded gene expression profiles of GSE53819 from the Gene Expression Omnibus (GEO) database, including 18 HNSC tissues and 18 normal tissues. -

GSTA3 Antibody
Product Datasheet GSTA3 Antibody Catalog No: #36515 Orders: [email protected] Description Support: [email protected] Product Name GSTA3 Antibody Host Species Rabbit Clonality Polyclonal Purification Antigen affinity purification. Applications WB IHC Species Reactivity Hu Specificity The antibody detects endogenous levels of total GSTA3 protein. Immunogen Type Recombinant Protein Immunogen Description Full length fusion protein Target Name GSTA3 Other Names GTA3; GSTA3-3 Accession No. Swiss-Prot#: Q16772NCBI Gene ID: 2940Gene Accssion: BC020619 SDS-PAGE MW 25kd Concentration 1mg/ml Formulation Rabbit IgG in pH7.4 PBS, 0.05% NaN3, 40% Glycerol. Storage Store at -20°C Application Details Western blotting: 1:500-1:2000 Immunohistochemistry: 1:50-1:200 Images Gel: 10%SDS-PAGE Lysates (from left to right): Human testis tissue and HepG2 cell Amount of lysate: 40ug per lane Primary antibody: 1/250 dilution Secondary antibody dilution: 1/8000 Exposure time: 10 seconds Address: 8400 Baltimore Ave., Suite 302, College Park, MD 20740, USA http://www.sabbiotech.com 1 Immunohistochemical analysis of paraffin-embedded Human colon cancer tissue using #36515 at dilution 1/30. Background Cytosolic and membrane-bound forms of glutathione S-transferase are encoded by two distinct supergene families. These enzymes are involved in cellular defense against toxic, carcinogenic, and pharmacologically active electrophilic compounds. At present, eight distinct classes of the soluble cytoplasmic mammalian glutathione S-transferases have been identified: alpha, kappa, mu, omega, pi, sigma, theta and zeta. This gene encodes a glutathione S-tranferase belonging to the alpha class genes that are located in a cluster mapped to chromosome 6. Genes of the alpha class are highly related and encode enzymes with glutathione peroxidase activity. -

GSTA3 Antibody (Monoclonal) (M01) Mouse Monoclonal Antibody Raised Against a Full Length Recombinant GSTA3
10320 Camino Santa Fe, Suite G San Diego, CA 92121 Tel: 858.875.1900 Fax: 858.622.0609 GSTA3 Antibody (monoclonal) (M01) Mouse monoclonal antibody raised against a full length recombinant GSTA3. Catalog # AT2277a Specification GSTA3 Antibody (monoclonal) (M01) - Product Information Application WB, IHC, E Primary Accession Q16772 Other Accession BC020619 Reactivity Human Host mouse Clonality Monoclonal Isotype IgG2a Kappa Calculated MW 25302 GSTA3 Antibody (monoclonal) (M01) - Additional Information Antibody Reactive Against Recombinant Protein.Western Blot detection against Gene ID 2940 Immunogen (50.16 KDa) . Other Names Glutathione S-transferase A3, GST class-alpha member 3, Glutathione S-transferase A3-3, GSTA3 Target/Specificity GSTA3 (AAH20619, 1 a.a. ~ 222 a.a) full-length recombinant protein with GST tag. MW of the GST tag alone is 26 KDa. Dilution WB~~1:500~1000 Format Clear, colorless solution in phosphate GSTA3 monoclonal antibody (M01), clone buffered saline, pH 7.2 . 1F11 Western Blot analysis of GSTA3 expression in HepG2 ( (Cat # AT2277a ) Storage Store at -20°C or lower. Aliquot to avoid repeated freezing and thawing. Precautions GSTA3 Antibody (monoclonal) (M01) is for research use only and not for use in diagnostic or therapeutic procedures. GSTA3 Antibody (monoclonal) (M01) - Protocols Page 1/3 10320 Camino Santa Fe, Suite G San Diego, CA 92121 Tel: 858.875.1900 Fax: 858.622.0609 Provided below are standard protocols that you may find useful for product applications. • Western Blot • Blocking Peptides • Dot Blot • Immunohistochemistry • Immunofluorescence • Immunoprecipitation • Flow Cytomety • Cell Culture Immunoperoxidase of monoclonal antibody to GSTA3 on formalin-fixed paraffin-embedded human tonsil. [antibody concentration 3 ug/ml] Detection limit for recombinant GST tagged GSTA3 is approximately 0.3ng/ml as a capture antibody. -

Snps in Genes Coding for ROS Metabolism and Signalling in Association with Docetaxel Clearance
The Pharmacogenomics Journal (2010) 10, 513–523 & 2010 Macmillan Publishers Limited. All rights reserved 1470-269X/10 www.nature.com/tpj ORIGINAL ARTICLE SNPs in genes coding for ROS metabolism and signalling in association with docetaxel clearance H Edvardsen1,2, PF Brunsvig3, The dose of docetaxel is currently calculated based on body surface area 1,4 5 and does not reflect the pharmacokinetic, metabolic potential or genetic H Solvang , A Tsalenko , background of the patients. The influence of genetic variation on the 6 7 A Andersen , A-C Syvanen , clearance of docetaxel was analysed in a two-stage analysis. In step one, 583 Z Yakhini5, A-L Børresen-Dale1,2, single-nucleotide polymorphisms (SNPs) in 203 genes were genotyped on H Olsen6, S Aamdal3 and samples from 24 patients with locally advanced non-small cell lung cancer. 1,2 We found that many of the genes harbour several SNPs associated with VN Kristensen clearance of docetaxel. Most notably these were four SNPs in EGF, three SNPs 1Department of Genetics, Institute of Cancer in PRDX4 and XPC, and two SNPs in GSTA4, TGFBR2, TNFAIP2, BCL2, DPYD Research, Oslo University Hospital Radiumhospitalet, and EGFR. The multiple SNPs per gene suggested the existence of common Oslo, Norway; 2Institute of Clinical Medicine, haplotypes associated with clearance. These were confirmed with detailed 3 University of Oslo, Oslo, Norway; Cancer Clinic, haplotype analysis. On the basis of analysis of variance (ANOVA), quantitative Oslo University Hospital Radiumhospitalet, Oslo, Norway; 4Institute of -
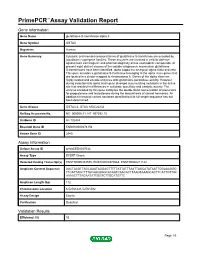
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name glutathione S-transferase alpha 3 Gene Symbol GSTA3 Organism Human Gene Summary Cytosolic and membrane-bound forms of glutathione S-transferase are encoded by two distinct supergene families. These enzymes are involved in cellular defense against toxic carcinogenic and pharmacologically active electrophilic compounds. At present eight distinct classes of the soluble cytoplasmic mammalian glutathione S-transferases have been identified: alpha kappa mu omega pi sigma theta and zeta. This gene encodes a glutathione S-tranferase belonging to the alpha class genes that are located in a cluster mapped to chromosome 6. Genes of the alpha class are highly related and encode enzymes with glutathione peroxidase activity. However during evolution this alpha class gene diverged accumulating mutations in the active site that resulted in differences in substrate specificity and catalytic activity. The enzyme encoded by this gene catalyzes the double bond isomerization of precursors for progesterone and testosterone during the biosynthesis of steroid hormones. An additional transcript variant has been identified but its full length sequence has not been determined. Gene Aliases GSTA3-3, GTA3, MGC22232 RefSeq Accession No. NC_000006.11, NT_007592.15 UniGene ID Hs.102484 Ensembl Gene ID ENSG00000174156 Entrez Gene ID 2940 Assay Information Unique Assay ID qHsaCED0047032 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000334575, ENST00000370968, ENST00000211122 Amplicon Context Sequence AACTAAGTTAGCAAATAGGAGTTTTTATTATTTAATTAGCATATAATTGGAAAGGG TTCATTAGCTTTACAACAGGCACAATCAACACTTAAGTAAAGCACTTCATTGTTGC AAAACTTTAGAATATTGGTCTTGCATGTTC Amplicon Length (bp) 112 Chromosome Location 6:52761441-52761582 Assay Design Exonic Purification Desalted Validation Results Efficiency (%) 93 Page 1/5 PrimePCR™Assay Validation Report R2 0.9991 cDNA Cq 34.67 cDNA Tm (Celsius) 76.5 gDNA Cq 24.84 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. -

Identification of Novel Regulatory Genes in Acetaminophen
IDENTIFICATION OF NOVEL REGULATORY GENES IN ACETAMINOPHEN INDUCED HEPATOCYTE TOXICITY BY A GENOME-WIDE CRISPR/CAS9 SCREEN A THESIS IN Cell Biology and Biophysics and Bioinformatics Presented to the Faculty of the University of Missouri-Kansas City in partial fulfillment of the requirements for the degree DOCTOR OF PHILOSOPHY By KATHERINE ANNE SHORTT B.S, Indiana University, Bloomington, 2011 M.S, University of Missouri, Kansas City, 2014 Kansas City, Missouri 2018 © 2018 Katherine Shortt All Rights Reserved IDENTIFICATION OF NOVEL REGULATORY GENES IN ACETAMINOPHEN INDUCED HEPATOCYTE TOXICITY BY A GENOME-WIDE CRISPR/CAS9 SCREEN Katherine Anne Shortt, Candidate for the Doctor of Philosophy degree, University of Missouri-Kansas City, 2018 ABSTRACT Acetaminophen (APAP) is a commonly used analgesic responsible for over 56,000 overdose-related emergency room visits annually. A long asymptomatic period and limited treatment options result in a high rate of liver failure, generally resulting in either organ transplant or mortality. The underlying molecular mechanisms of injury are not well understood and effective therapy is limited. Identification of previously unknown genetic risk factors would provide new mechanistic insights and new therapeutic targets for APAP induced hepatocyte toxicity or liver injury. This study used a genome-wide CRISPR/Cas9 screen to evaluate genes that are protective against or cause susceptibility to APAP-induced liver injury. HuH7 human hepatocellular carcinoma cells containing CRISPR/Cas9 gene knockouts were treated with 15mM APAP for 30 minutes to 4 days. A gene expression profile was developed based on the 1) top screening hits, 2) overlap with gene expression data of APAP overdosed human patients, and 3) biological interpretation including assessment of known and suspected iii APAP-associated genes and their therapeutic potential, predicted affected biological pathways, and functionally validated candidate genes. -

Elucidation of Melanogenesis-Associated Signaling Pathways Regulated by Argan Press Cake in B16 Melanoma Cells
nutrients Article Elucidation of Melanogenesis-Associated Signaling Pathways Regulated by Argan Press Cake in B16 Melanoma Cells Thouria Bourhim 1,‡ , Myra O. Villareal 2,3,‡ , Chemseddoha Gadhi 1,2,* and Hiroko Isoda 2,3,* 1 Faculty of Sciences Semlalia, Cadi Ayyad University, Avenue Prince Moulay Abdellah, B.P. 2390, Marrakesh 40000, Morocco; [email protected] 2 Alliance for Research on the Mediterranean and North Africa (ARENA), University of Tsukuba, Tennodai 1-1-1, Tsukuba 305-8572, Japan; [email protected] 3 Faculty of Life and Environmental Sciences, University of Tsukuba, Tennodai 1-1-1, Tsukuba 305-8572, Japan * Correspondence: [email protected] (C.G.); [email protected] (H.I.) ‡ These authors contributed equally to this work. Abstract: The beneficial effect on health of argan oil is recognized worldwide. We have previously reported that the cake that remains after argan oil extraction (argan press-cake or APC) inhibits melanogenesis in B16 melanoma cells in a time-dependent manner without cytotoxicity. In this study, the global gene expression profile of B16 melanoma cells treated with APC extract was determined in order to gain an understanding of the possible mechanisms of action of APC. The results suggest that APC extract inhibits melanin biosynthesis by down-regulating microphthalmia- associated transcription factor (Mitf ) and its downstream signaling pathway through JNK signaling activation, and the inhibition of Wnt/β-catenin and cAMP/PKA signaling pathways. APC extract also prevented the transport of melanosomes by down-regulating Rab27a expression. These results suggest that APC may be an important natural skin whitening product and pharmacological agent used for clinical treatment of pigmentary disorders. -
Identification of Genes and Signaling Pathways Associated with Squamous Cell Carcinoma by Bioinformatics Analysis
1382 ONCOLOGY LETTERS 11: 1382-1390, 2016 Identification of genes and signaling pathways associated with squamous cell carcinoma by bioinformatics analysis LI SHEN1,2, LINBO LIU1, ZHENYONG YANG2 and NAN JIANG2 1Department of Plastic Surgery, The First Affiliated Hospital of Zhengzhou University; 2Department of Plastic and Aesthetic Surgery, The Fifth Affiliated Hospital of Zhengzhou University, Zhengzhou, Henan 450052, P.R. China Received December 18, 2014; Accepted November 2, 2015 DOI: 10.3892/ol.2015.4051 Abstract. The present study aimed to investigate the genes Introduction and signaling pathways associated with squamous cell carci- noma (SCC) by bioinformatics analysis. For this purpose, Squamous cell carcinoma (SCC) is a histologically distinct the GSE2503 was downloaded from the Gene Expression type of cancer (1). It arises from the uncontrolled prolifera- Omnibus database, and the differentially expressed genes tion of epithelial cells or cells exhibiting cytological or tissue (DEGs) between 6 normal skin and 5 SCC samples were architectural characteristics of SC differentiation, including analyzed using the Linear Models for Microarray Data the presence of keratin, tonofilament bundles or desmosomes, package. Gene Ontology (GO) and pathway enrichment which are structures involved in cell-cell adhesion (1). SCC analysis of DEGs were performed, followed by functional occurs in numerous tissues, including the lips, mouth, esoph- annotation and construction of a protein-protein interac- agus, lungs, urinary bladder and prostate (2). Of all the cases tion (PPI) network. Subnetwork modules were subsequently of SCC, 2.5% become metastatic and lead to substantial identified and analyzed. A total of 181 DEGs, including morbidity, which constitutes a considerable economic burden 95 upregulated and 86 downregulated DEGs, were identified, to the healthcare system (3).