Phylogenetic and Functional Diversity of Soil Prokaryotic Communities in Temperate Deciduous Forests with Different Tree Species
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Novel Bacterial Lineages Associated with Boreal Moss Species Hannah
bioRxiv preprint doi: https://doi.org/10.1101/219659; this version posted November 16, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 Novel bacterial lineages associated with boreal moss species 2 Hannah Holland-Moritz1,2*, Julia Stuart3, Lily R. Lewis4, Samantha Miller3, Michelle C. Mack3, Stuart 3 F. McDaniel4, Noah Fierer1,2* 4 Affiliations: 5 1Cooperative Institute for Research in Environmental Sciences, University of Colorado at Boulder, 6 Boulder, CO, USA 7 2Department of Ecology and Evolutionary Biology, University of Colorado at Boulder, Boulder, CO, 8 USA 9 3Center for Ecosystem Science and Society, Northern Arizona University, Flagstaff, AZ USA 10 4Department of Biology, University of Florida, Gainesville, FL 32611-8525, USA 11 *Corresponding Author 12 13 Abstract 14 Mosses are critical components of boreal ecosystems where they typically account for a large 15 proportion of net primary productivity and harbor diverse bacterial communities that can be the major 16 source of biologically-fixed nitrogen in these ecosystems. Despite their ecological importance, we have 17 limited understanding of how microbial communities vary across boreal moss species and the extent to 18 which local environmental conditions may influence the composition of these bacterial communities. 19 We used marker gene sequencing to analyze bacterial communities associated with eight boreal moss 20 species collected near Fairbanks, AK USA. We found that host identity was more important than site in 21 determining bacterial community composition and that mosses harbor diverse lineages of potential N2- 22 fixers as well as an abundance of novel taxa assigned to understudied bacterial phyla (including 23 candidate phylum WPS-2). -

Managing Midas
Managing MIDAs Managing MIDAs Harmonising the management of Multi-Internationally Designated Areas: Ramsar Sites, World Heritage sites, Biosphere Reserves and UNESCO Global Geoparks Thomas Schaaf and Diana Clamote Rodrigues INTERNATIONAL UNION FOR CONSERVATION OF NATURE WORLD HEADQUARTERS Rue Mauverney 28 1196 Gland, Switzerland Tel +41 22 999 0000 Fax +41 22 999 0002 www.iucn.org IUCN IUCN (International Union for Conservation of Nature) IUCN is a membership Union composed of both government and civil society organisations. It harnesses the experience, resources and reach of its more than 1,300 Member organisations and the input of more than 16,000 experts. IUCN is the global authority on the status of the natural world and the measures needed to safeguard it. www.iucn.org Ramsar Convention The Convention on Wetlands, called the Ramsar Convention, is an intergovernmental treaty that provides the framework for national action and international cooperation for the conservation and wise use of wetlands and their resources. Its mission is “the conservation and wise use of all wetlands through local and national actions and international cooperation, as a contribution towards achieving sustainable development throughout the world”. Under the “three pillars” of the Convention, the Contracting Parties commit to: work towards the wise use of all their wetlands; designate suitable wetlands for the list of Wetlands of International Importance (the “Ramsar List”) and ensure their effective management; and cooperate internationally on transboundary wetlands, shared wetland systems and shared species. www.ramsar.org NIO M O UN IM D R T IA A L • World Heritage Convention P • W L O A I R D L D N H O E M R I E TA IN G O The 1972 Convention concerning the Protection of the World Cultural and Natural Heritage recognises that certain E • PATRIM United Nations World Educational, Scientific and Heritage places on Earth are of “outstanding universal value” and should form part of the common heritage of humankind. -

Genomic Analysis of Family UBA6911 (Group 18 Acidobacteria)
bioRxiv preprint doi: https://doi.org/10.1101/2021.04.09.439258; this version posted April 10, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. 1 2 Genomic analysis of family UBA6911 (Group 18 3 Acidobacteria) expands the metabolic capacities of the 4 phylum and highlights adaptations to terrestrial habitats. 5 6 Archana Yadav1, Jenna C. Borrelli1, Mostafa S. Elshahed1, and Noha H. Youssef1* 7 8 1Department of Microbiology and Molecular Genetics, Oklahoma State University, Stillwater, 9 OK 10 *Correspondence: Noha H. Youssef: [email protected] bioRxiv preprint doi: https://doi.org/10.1101/2021.04.09.439258; this version posted April 10, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. 11 Abstract 12 Approaches for recovering and analyzing genomes belonging to novel, hitherto unexplored 13 bacterial lineages have provided invaluable insights into the metabolic capabilities and 14 ecological roles of yet-uncultured taxa. The phylum Acidobacteria is one of the most prevalent 15 and ecologically successful lineages on earth yet, currently, multiple lineages within this phylum 16 remain unexplored. Here, we utilize genomes recovered from Zodletone spring, an anaerobic 17 sulfide and sulfur-rich spring in southwestern Oklahoma, as well as from multiple disparate soil 18 and non-soil habitats, to examine the metabolic capabilities and ecological role of members of 19 the family UBA6911 (group18) Acidobacteria. -

The Archaea Community Associated with Lava-Formed Gotjawal Forest Soil in Jeju, Korea
Journal of Agricultural Chemistry and Environment, 2014, 3, 96-102 Published Online August 2014 in SciRes. http://www.scirp.org/journal/jacen http://dx.doi.org/10.4236/jacen.2014.33012 The Archaea Community Associated with Lava-Formed Gotjawal Forest Soil in Jeju, Korea Jong-Shik Kim1*, Man-Young Jung2, Keun Chul Lee3, Dae-Shin Kim4, Suk-Hyung Ko4, Jung-Sook Lee3, Sung-Keun Rhee2 1Gyeongbuk Institute for Marine Bioindustry, Uljin, Republic of Korea 2Department of Microbiology, Chungbuk National University, Cheongju, Republic of Korea 3Korea Research Institute of Bioscience and Biotechnology, Daejeon, Republic of Korea 4Research Institute for Hallasan, Jeju, Republic of Korea Email: *[email protected] Received 22 April 2014; revised 27 May 2014; accepted 23 June 2014 Copyright © 2014 by authors and Scientific Research Publishing Inc. This work is licensed under the Creative Commons Attribution International License (CC BY). http://creativecommons.org/licenses/by/4.0/ Abstract The abundance and diversity of archaeal assemblages were analyzed in soils collected from Gyo- rae Gotjawal forest, Jeju, Korea. Gotjawal soil refers to soil derived from a lava-formed forest, cha- racterized by high organic matter content, fertility, and poor rocky soil. Using domain-specific primers, archaeal 16S rRNA gene sequences were PCR amplified for clone library construction, and a total of 185 archaeal clones were examined. The archaeal clones were affiliated with the phyla Thaumarchaeota (96.2%) and Euryarchaeota (3.8%). The most abundant thaumarchaeal group (90.3% of the clones) was the group I.1b clade, which includes soil ammonia-oxidizing arc- haea. The unique characteristics of Gotjawal soil, including basalt morphology, vegetation, and groundwater aquifer penetration, may be reflected in the archaeal community composition. -
Benefit What Is the JEJU MICE CARD? JEJU MICE CARD란?
Benefit What is the JEJU MICE CARD? JEJU MICE CARD란? JEJU MICE CARD is a trans portation card that makes it easy to use bus (including re gular bus, limousine bus, City 12/18 Tour Bus, etc) on Jeju Island. You can comfortably recharge the card at all of theses con venience stores(GS25, Seven Eleven, CU). JEJU MICE CARD offers all card users up to 60% discount and other benefits related to all affiliated company such as various tourist attractions, hotels, restaurants, shopping stores, etc. JEJU MICE 카드는 제주도내 시내·외 버스 및 리무진, 시티투어버스 이용시 교통 카드로 사용가능하며, GS25, 세븐일레븐, CU 편의점에서 결제가능합니다. 또한, 제주도내 관광지, 체험프로그램, 맛집, 호텔 레스토랑 등 다양한 제휴 가맹점에서 최대 60%할인 혜택과 우대서비스를 제공합니다. * It’s charged 5,000 won at first. * 기본 5,000원이 충전되어 있습니다. Transportation card 교통카드 기능 You can use it when you use the transportation. 일반버스 및 리무진, 시티투어 버스 등 버스 이용시 교통카드로 사용 가능합니다. Easy recharging and payment at CVS 편의점에서 충전 및 결제가능 You can recharge it at CVS in Jeju easily. 제주도내 편의점에서 쉽게 충전하여 사용할 수 있습니다. How to Charge 충전방법 Cash recharging only 현금충전만 가능 편의점(CVS) * You can recharge JEJU MICE CARD at CVS(SEVEN ELEVEN, GS25, CU) 제주도내 편의점에서 충전가능합니다. How to use the JEJU MICE card 카드 사용방법 When you use the transportation 대중교통 이용 Touch Payment 단말기 접촉 * Must touch the card to the card reader when get on and get off * 승/하차시 반드시 태그해야 함 Affiliated JEJUPASS member stores DC 제휴 가맹점 할인 Present your card for Discount 제주마이스카드 제시 후 할인 You can get a 10% discount when you show JEJU MICE Card at the stores in ICC JEJU. -

Tree-Aggregated Predictive Modeling of Microbiome Data
www.nature.com/scientificreports OPEN Tree‑aggregated predictive modeling of microbiome data Jacob Bien1, Xiaohan Yan2, Léo Simpson3,4 & Christian L. Müller4,5,6* Modern high‑throughput sequencing technologies provide low‑cost microbiome survey data across all habitats of life at unprecedented scale. At the most granular level, the primary data consist of sparse counts of amplicon sequence variants or operational taxonomic units that are associated with taxonomic and phylogenetic group information. In this contribution, we leverage the hierarchical structure of amplicon data and propose a data‑driven and scalable tree‑guided aggregation framework to associate microbial subcompositions with response variables of interest. The excess number of zero or low count measurements at the read level forces traditional microbiome data analysis workfows to remove rare sequencing variants or group them by a fxed taxonomic rank, such as genus or phylum, or by phylogenetic similarity. By contrast, our framework, which we call trac (tree‑aggregation of compositional data), learns data‑adaptive taxon aggregation levels for predictive modeling, greatly reducing the need for user‑defned aggregation in preprocessing while simultaneously integrating seamlessly into the compositional data analysis framework. We illustrate the versatility of our framework in the context of large‑scale regression problems in human gut, soil, and marine microbial ecosystems. We posit that the inferred aggregation levels provide highly interpretable taxon groupings that can help microbiome researchers gain insights into the structure and functioning of the underlying ecosystem of interest. Microbial communities populate all major environments on earth and signifcantly contribute to the total plan- etary biomass. Current estimates suggest that a typical human-associated microbiome consists of ∼ 1013 bacteria1 and that marine bacteria and protists contribute to as much as 70% of the total marine biomass2. -

Motions Issued on 8 July 2012
Congress Document WCC-2012-9.6* Motions Issued on 8 July 2012 World Conservation Congress, Jeju, Republic of Korea 6–15 September 2012 *This document is also submitted for agenda items 1.8, 2.1.6, 3.1.6, 4.1.6 and 6.1.6. Table of Contents Title Categories 001 Strengthening the motions process and enhancing implementation of IUCN Resolutions IUCN Governance 002 Improved opportunity for Member participation in IUCN IUCN Governance 003 Prioritizing IUCN membership awareness and support IUCN Governance 004 Establishment of the Ethics Mechanism IUCN Governance Strengthening of the IUCN National and Regional Committees and the optional use of the 005 three official languages in documents for internal and external communication by IUCN IUCN Governance and its Members Cooperation with regional government authorities in the implementation of the IUCN 006 IUCN Governance Programme 2013–2016 Establishing an Indigenous Peoples’ Organization (IPO) membership and voting category 007 IUCN Governance in IUCN Increasing youth engagement and intergenerational partnership across and through the 008 IUCN Governance Union 009 Encouraging cooperation with faith-based organizations and networks IUCN Governance 010 Establishment of a strengthened institutional presence of IUCN in North-East Asia IUCN Governance 011 Consolidating IUCN’s institutional presence in South America IUCN Governance 012 Strengthening IUCN in the insular Caribbean IUCN Governance 013 IUCN’s name IUCN Governance 014 Implementing Aichi Target 12 of the Strategic Plan for Biodiversity 2011–2020 -

Human Influence, Regeneration, and Conservation of the Gotjawal Forests
Journal of Marine and Island Cultures (2013) 2, 85–92 Journal of Marine and Island Cultures www.sciencedirect.com Human influence, regeneration, and conservation of the Gotjawal forests in Jeju Island, Korea Hong-Gu Kang a,*, Chan-Soo Kim b, Eun-Shik Kim c,* a Department of Forest Resources, Graduate School, Kookmin University, Seoul 136-702, Republic of Korea b Warm-Temperate and Subtropical Forest Research Center, Korea Forest Research Institute, Jeju 697-050, Republic of Korea c Department of Forestry, Environment, and Systems, Kookmin University, Seoul 136-702, Republic of Korea Received 29 October 2013 Available online 5 December 2013 KEYWORDS Abstract Gotjawal, a uniquely formed forest vegetation on the lava terrain located at eastern and Conservation; western parts of Jeju Island, covers 6% of the island’s land surface. The Gotjawal forests play Ecosystem services; important roles in establishing the biological and cultural diversity while maintaining ecosystem ser- Gotjawal forests; vices. Recently, with the recognition of the diverse ecological and cultural values of the Gotjawal Human influence; forests, efforts to conserve the forests were conducted by adopting the resolutions of the Jeju World Jeju Island; Conservation Congress of the IUCN held in 2012. Despite its precious values, the Gotjawal forest is Regeneration being threatened by the developmental activities of large scale constructions projects. To under- stand the recent regeneration of the Gotjawal forests, ecological studies have been conducted at the Hankyeong-Andeok Gotjawal Terrain, which is located in the western part and occupies the largest area of the Gotjawal Terrain of Jeju Island. Major vegetation in the area includes the decid- uous broad-leaved forests (Acer palmatum–Styrax japonicus community), mixed deciduous and evergreen broad-leaved forests (Neolitsea aciculata–Styrax japonicus community), and evergreen broad-leaved forests (Quercus glauca community). -

Microbial Communities of Polymetallic Deposits' Acidic
Microbial Communities of Polymetallic Deposits’ Acidic Ecosystems of ANGOR UNIVERSITY Continental Climatic Zone With High Temperature Contrasts Gavrilov, Sergei N.; Korzhenkov, Aleksei A.; Kublanov, Ilya V.; Bargiela, Rafael; Zamana, Leonid V.; Toshchakov, Stepan V.; Golyshin, Peter; Golyshina, Olga Frontiers in Microbiology PRIFYSGOL BANGOR / B Published: 17/07/2019 Publisher's PDF, also known as Version of record Cyswllt i'r cyhoeddiad / Link to publication Dyfyniad o'r fersiwn a gyhoeddwyd / Citation for published version (APA): Gavrilov, S. N., Korzhenkov, A. A., Kublanov, I. V., Bargiela, R., Zamana, L. V., Toshchakov, S. V., Golyshin, P., & Golyshina, O. (2019). Microbial Communities of Polymetallic Deposits’ Acidic Ecosystems of Continental Climatic Zone With High Temperature Contrasts. Frontiers in Microbiology. Hawliau Cyffredinol / General rights Copyright and moral rights for the publications made accessible in the public portal are retained by the authors and/or other copyright owners and it is a condition of accessing publications that users recognise and abide by the legal requirements associated with these rights. • Users may download and print one copy of any publication from the public portal for the purpose of private study or research. • You may not further distribute the material or use it for any profit-making activity or commercial gain • You may freely distribute the URL identifying the publication in the public portal ? Take down policy If you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediately and investigate your claim. 01. Oct. 2021 ORIGINAL RESEARCH published: 17 July 2019 doi: 10.3389/fmicb.2019.01573 Microbial Communities of Polymetallic Deposits’ Acidic Ecosystems of Continental Climatic Zone With High Temperature Contrasts Sergey N. -
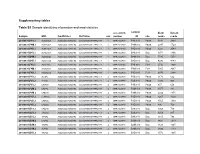
Supplementary Tables Table S1 Sample Identifying Information And
Supplementary tables Table S1 Sample identifying information and read statistics accession sample #raw #clean Sample MID FwdPrimer RvPrimer run number ID site reads reads 20100831.P4S.1 ACACGAGA ACGGGCGGTGWGTRC CCGYCAATTCMTTTRAGTTT 1 SRR1023739 P4S.910 Palsa 3831 2893 20100831.P4M.1 ACATAGCA ACGGGCGGTGWGTRC CCGYCAATTCMTTTRAGTTT 1 SRR1023739 P4M.910 Palsa 2047 1729 20100831.P4D.1 ACGATACA ACGGGCGGTGWGTRC CCGYCAATTCMTTTRAGTTT 1 SRR1023739 P4D.910 Palsa 3221 2543 20100831.B4S.1 ACTGCTCA ACGGGCGGTGWGTRC CCGYCAATTCMTTTRAGTTT 1 SRR1023739 B4S.910 Bog 1971 1366 20100831.B4M.1 AGCAGCGC ACGGGCGGTGWGTRC CCGYCAATTCMTTTRAGTTT 1 SRR1023739 B4M.910 Bog 5530 5251 20100831.B4D.1 AGCGTGCA ACGGGCGGTGWGTRC CCGYCAATTCMTTTRAGTTT 1 SRR1023739 B4D.910 Bog 4296 4143 20100831.F4S.1 AGTCTAGC ACGGGCGGTGWGTRC CCGYCAATTCMTTTRAGTTT 1 SRR1023739 F4S.910 Fen* 2716 1890 20100831.F4M.1 ATGACTGA ACGGGCGGTGWGTRC CCGYCAATTCMTTTRAGTTT 1 SRR1023739 F4M.910 Fen* 5005 3987 20100831.F4D.1 ATGCACGC ACGGGCGGTGWGTRC CCGYCAATTCMTTTRAGTTT 1 SRR1023739 F4D.910 Fen* 2870 2384 20100901.P1S.2 ACTGAT ACGGGCGGTGWGTRC CCGYCAATTCMTTTRAGTTT 2 SRR1023740 P1S.910 Palsa 1170 662 20100901.P2S.2 ATGTGT ACGGGCGGTGWGTRC CCGYCAATTCMTTTRAGTTT 2 SRR1023740 P2S.910 Palsa 1320 923 20100901.P3S.2 CACAGT ACGGGCGGTGWGTRC CCGYCAATTCMTTTRAGTTT 2 SRR1023740 P3S.910 Palsa 857 656 20100901.P2M.2 CACTAC ACGGGCGGTGWGTRC CCGYCAATTCMTTTRAGTTT 2 SRR1023740 P2M.910 Palsa 1077 881 20100901.P3M.2 CAGCAT ACGGGCGGTGWGTRC CCGYCAATTCMTTTRAGTTT 2 SRR1023740 P3M.910 Palsa 2032 1474 20100901.P1D.2 CTAGAGC ACGGGCGGTGWGTRC CCGYCAATTCMTTTRAGTTT -

Pyrosequencing Investigation Into the Bacterial Community in Permafrost Soils Along the China-Russia Crude Oil Pipeline (CRCOP)
Pyrosequencing Investigation into the Bacterial Community in Permafrost Soils along the China-Russia Crude Oil Pipeline (CRCOP) Sizhong Yang1*, Xi Wen2, Huijun Jin1, Qingbai Wu1 1 State Key Laboratory of Frozen Soil Engineering (SKLFSE), Cold and Arid Regions Environmental and Engineering Research Institute (CAREERI), Chinese Academy of Sciences, Lanzhou, China, 2 College of Electrical Engineering, Northwest University for Nationalities, Lanzhou, China Abstract The China-Russia Crude Oil Pipeline (CRCOP) goes through 441 km permafrost soils in northeastern China. The bioremediation in case of oil spills is a major concern. So far, little is known about the indigenous bacteria inhabiting in the permafrost soils along the pipeline. A pilot 454 pyrosequencing analysis on the communities from four selected sites which possess high environment risk along the CRCOP is herein presented. The results reveal an immense bacterial diversity than previously anticipated. A total of 14448 OTUs with 84834 reads are identified, which could be assigned into 39 different phyla, and 223 families or 386 genera. Only five phyla sustain a mean OTU abundance more than 5% in all the samples, but they altogether account for 85.08% of total reads. Proteobacteria accounts for 41.65% of the total OTUs or 45% of the reads across all samples, and its proportion generally increases with soil depth, but OTUs numerically decline. Among Proteobacteria, the abundance of Beta-, Alpha-, Delta- and Gamma- subdivisions average to 38.7% (2331 OTUs), 37.5% (2257 OTUs), 10.35% (616 OTUs), and 6.21% (374 OTUs), respectively. Acidobacteria (esp. Acidobacteriaceae), Actinobacteria (esp. Intrasporangiaceae), Bacteroidetes (esp. Sphingobacteria and Flavobacteria) and Chloroflexi (esp. -

A Comparison of Computationally Predicted Functional Metagenomes and Microarray Analysis for Microbial P Cycle Genes in a Unique
F1000Research 2018, 7:179 Last updated: 31 JUL 2021 BRIEF REPORT A comparison of computationally predicted functional metagenomes and microarray analysis for microbial P cycle genes in a unique basalt-soil forest [version 1; peer review: 2 approved] Erick S. LeBrun , Sanghoon Kang Center for Reservoir and Aquatic Systems Research, Department of Biology, Baylor University, Waco, TX, 76798-7388, USA v1 First published: 12 Feb 2018, 7:179 Open Peer Review https://doi.org/10.12688/f1000research.13841.1 Latest published: 12 Feb 2018, 7:179 https://doi.org/10.12688/f1000research.13841.1 Reviewer Status Invited Reviewers Abstract Here we compared microbial results for the same Phosphorus (P) 1 2 biogeochemical cycle genes from a GeoChip microarray and PICRUSt functional predictions from 16S rRNA data for 20 samples in the four version 1 spatially separated Gotjawal forests on Jeju Island in South Korea. The 12 Feb 2018 report report high homogeneity of microbial communities detected at each site allows sites to act as environmental replicates for comparing the two 1. Ye Deng, Chinese Academy of Sciences, different functional analysis methods. We found that while both methods capture the homogeneity of the system, both differed Beijing, China greatly in the total abundance of genes detected, as well as the 2. Joy D Van Nostrand , University of diversity of taxa detected. Additionally, we introduce a more comprehensive functional assay that again captures the homogeneity Oklahoma, Norman, USA of the system but also captures more extensive community gene and taxonomic information and depth. While both methods have their Any reports and responses or comments on the advantages and limitations, PICRUSt appears better suited to asking article can be found at the end of the article.