Facilitating Complex Trait Analysis Via Reduced Complexity Crosses
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Lineage-Specific Mapping of Quantitative Trait Loci
Heredity (2013) 111, 106–113 & 2013 Macmillan Publishers Limited All rights reserved 0018-067X/13 www.nature.com/hdy ORIGINAL ARTICLE Lineage-specific mapping of quantitative trait loci C Chen1 and K Ritland We present an approach for quantitative trait locus (QTL) mapping, termed as ‘lineage-specific QTL mapping’, for inferring allelic changes of QTL evolution along with branches in a phylogeny. We describe and analyze the simplest case: by adding a third taxon into the normal procedure of QTL mapping between pairs of taxa, such inferences can be made along lineages to a presumed common ancestor. Although comparisons of QTL maps among species can identify homology of QTLs by apparent co- location, lineage-specific mapping of QTL can classify homology into (1) orthology (shared origin of QTL) versus (2) paralogy (independent origin of QTL within resolution of map distance). In this light, we present a graphical method that identifies six modes of QTL evolution in a three taxon comparison. We then apply our model to map lineage-specific QTLs for inbreeding among three taxa of yellow monkey-flower: Mimulus guttatus and two inbreeders M. platycalyx and M. micranthus, but critically assuming outcrossing was the ancestral state. The two most common modes of homology across traits were orthologous (shared ancestry of mutation for QTL alleles). The outbreeder M. guttatus had the fewest lineage-specific QTL, in accordance with the presumed ancestry of outbreeding. Extensions of lineage-specific QTL mapping to other types of data and crosses, and to inference of ancestral QTL state, are discussed. Heredity (2013) 111, 106–113; doi:10.1038/hdy.2013.24; published online 24 April 2013 Keywords: Quantitative trait locus mapping; evolution of mating systems; Mimulus INTRODUCTION common ancestor of these two taxa, back to the common ancestor of Knowledge of the genetic architecture of complex traits can offer all three taxa and forwards again to the third taxon. -
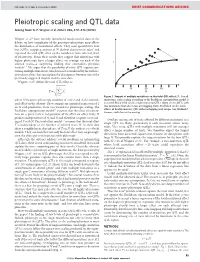
Pleiotropic Scaling and QTL Data Arising From: G
NATURE | Vol 456 | 4 December 2008 BRIEF COMMUNICATIONS ARISING Pleiotropic scaling and QTL data Arising from: G. P. Wagner et al. Nature 452, 470–473 (2008) Wagner et al.1 have recently introduced much-needed data to the debate on how complexity of the genotype–phenotype map affects the distribution of mutational effects. They used quantitative trait loci (QTLs) mapping analysis of 70 skeletal characters in mice2 and regressed the total QTL effect on the number of traits affected (level of pleiotropy). From their results they suggest that mutations with higher pleiotropy have a larger effect, on average, on each of the T affected traits—a surprising finding that contradicts previous models3–7. We argue that the possibility of some QTL regions con- taining multiple mutations, which was not considered by the authors, introduces a bias that can explain the discrepancy between one of the previously suggested models and the new data. Wagner et al.1 define the total QTL effect as vffiffiffiffiffiffiffiffiffiffiffiffiffi u uXN 10 20 30 ~t 2 T Ai N i~1 Figure 1 | Impact of multiple mutations on the total QTL effect, T. For all where N measures pleiotropy (number of traits) and Ai the normal- mutations, strict scaling according to the Euclidean superposition model is ized effect on the ith trait. They compare an empirical regression of T assumed. Black filled circles, single-mutation QTLs. Open circles, QTLs with on N with predictions from two models for pleiotropic scaling. The two mutations that affect non-overlapping traits. Red filled circles, total Euclidean superposition model3,4 assumes that the effect of a muta- effects of double-mutant QTL with overlapping trait ranges (see Methods). -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

1 ROBERT PLOMIN on Behavioral Genetics Genetic Links Common
Genetic links • Background – Why has educational psychology ben so slow ROBERT PLOMIN to accept genetics? on Behavioral Genetics • Beyond nature vs. nurture – 3 findings from genetic research with far- reaching implications for education: October, 2004 The nature of nurture The abnormal is normal Generalist genes • Molecular Genetics Quantitative genetics: twin and adoption studies Common medical disorders Identical twins Fraternal twins (monozygotic, MZ) (dizygotic, DZ) 1 Twin concordances in TEDS at 7 years Twins Early Development Study (TEDS) for low reading and maths Perceptions of nature/nurture: % parents (N=1340) and teachers Beyond nature vs. nurture: (N=667) indicating that genes are at least as important as environment (Walker & Plomin, Ed. Psych., in press) The nature of nurture • Genetic influence on experience • Genotype-environment correlation – Children’s genetic propensities are correlated with their experiences passive evocative active 2 Beyond nature vs. nurture: Genotype-environment correlation The nature of nurture • Passive • Genotype-environment correlation – e.g., family background (SES) and school – Children’s genetic propensities are correlated with achievement their experiences passive • Evocative evocative – Children evoke experiences for genetic reasons Active • Active • Molecular genetics – Children actively create environments that foster – Find genes associated with learning experiences their genetic propensities Beyond nature vs. nurture: The one-gene one-disability (OGOD) model The abnormal is normal • Common learning disabilities are the quantitative extreme of the same genetic and environmental factors responsible for normal variation in learning abilities. • There are no common learning disabilities, just the extremes of normal variation. 3 OGOD (rare) and QTL (common) disorders The quantitative trait locus (QTL) model Beyond nature vs. -

Genome-Wide Rnai Screening Identifies Human Proteins with A
RESOURCES Genome-wide RNAi screening identifies human proteins with a regulatory function in the early secretory pathway Jeremy C. Simpson1,7, Brigitte Joggerst2, Vibor Laketa2, Fatima Verissimo2, Cihan Cetin2, Holger Erfle2,6, Mariana G. Bexiga1, Vasanth R. Singan1, Jean-Karim Hériché3, Beate Neumann3, Alvaro Mateos2, Jonathon Blake4, Stephanie Bechtel5, Vladimir Benes4, Stefan Wiemann5, Jan Ellenberg2,3 and Rainer Pepperkok2,7 The secretory pathway in mammalian cells has evolved to facilitate the transfer of cargo molecules to internal and cell surface membranes. Use of automated microscopy-based genome-wide RNA interference screens in cultured human cells allowed us to identify 554 proteins influencing secretion. Cloning, fluorescent-tagging and subcellular localization analysis of 179 of these proteins revealed that more than two-thirds localize to either the cytoplasm or membranes of the secretory and endocytic pathways. The depletion of 143 of them resulted in perturbations in the organization of the COPII and/or COPI vesicular coat complexes of the early secretory pathway, or the morphology of the Golgi complex. Network analyses revealed a so far unappreciated link between early secretory pathway function, small GTP-binding protein regulation, actin cytoskeleton organization and EGF-receptor-mediated signalling. This work provides an important resource for an integrative understanding of global cellular organization and regulation of the secretory pathway in mammalian cells. Within higher eukaryotic cells membrane traffic pathways connect the Extensive efforts over many years have revealed a significant number various membrane-bounded organelles, thereby ensuring that they of regulators associated with the secretory pathway. Early biochemical retain the correct complement of proteins and lipids to maintain approaches to identify individual machinery components have started cellular homeostasis. -

S41436-020-01011-X.Pdf
Zurich Open Repository and Archive University of Zurich Main Library Strickhofstrasse 39 CH-8057 Zurich www.zora.uzh.ch Year: 2020 New insights into the clinical and molecular spectrum of the novel CYFIP2-related neurodevelopmental disorder and impairment of the WRC-mediated actin dynamics Begemann, Anaïs ; Sticht, Heinrich ; Begtrup, Amber ; Vitobello, Antonio ; Faivre, Laurence ; Asadollahi, Reza ; Zweier, Markus ; Steindl, Katharina ; Rauch, Anita Abstract: PURPOSE A few de novo missense variants in the cytoplasmic FMRP-interacting protein 2 (CYFIP2) gene have recently been described as a novel cause of severe intellectual disability, seizures, and hypotonia in 18 individuals, with p.Arg87 substitutions in the majority. METHODS We assembled data from 19 newly identified and all 18 previously published individuals with CYFIP2 variants. By structural modeling and investigation of WAVE-regulatory complex (WRC)-mediated actin polymerization in six patient fibroblast lines we assessed the impact of CYFIP2 variants on the WRC. RESULTS Sixteenof 19 individuals harbor two previously described and 11 novel (likely) disease-associated missense variants. We report p.Asp724 as second mutational hotspot (4/19 cases). Genotype-phenotype correlation con- firms a consistently severe phenotype in p.Arg87 patients but a more variable phenotype inp.Asp724 and other substitutions. Three individuals with milder phenotypes carry putative loss-of-function vari- ants, which remain of unclear pathogenicity. Structural modeling predicted missense variants to disturb interactions within the WRC or impair CYFIP2 stability. Consistent with its role in WRC-mediated actin polymerization we substantiate aberrant regulation of the actin cytoskeleton in patient fibroblasts. CONCLUSION Our study expands the clinical and molecular spectrum of CYFIP2-related neurodevel- opmental disorder and provides evidence for aberrant WRC-mediated actin dynamics as contributing cellular pathomechanism. -

ATP-Binding and Hydrolysis in Inflammasome Activation
molecules Review ATP-Binding and Hydrolysis in Inflammasome Activation Christina F. Sandall, Bjoern K. Ziehr and Justin A. MacDonald * Department of Biochemistry & Molecular Biology, Cumming School of Medicine, University of Calgary, 3280 Hospital Drive NW, Calgary, AB T2N 4Z6, Canada; [email protected] (C.F.S.); [email protected] (B.K.Z.) * Correspondence: [email protected]; Tel.: +1-403-210-8433 Academic Editor: Massimo Bertinaria Received: 15 September 2020; Accepted: 3 October 2020; Published: 7 October 2020 Abstract: The prototypical model for NOD-like receptor (NLR) inflammasome assembly includes nucleotide-dependent activation of the NLR downstream of pathogen- or danger-associated molecular pattern (PAMP or DAMP) recognition, followed by nucleation of hetero-oligomeric platforms that lie upstream of inflammatory responses associated with innate immunity. As members of the STAND ATPases, the NLRs are generally thought to share a similar model of ATP-dependent activation and effect. However, recent observations have challenged this paradigm to reveal novel and complex biochemical processes to discern NLRs from other STAND proteins. In this review, we highlight past findings that identify the regulatory importance of conserved ATP-binding and hydrolysis motifs within the nucleotide-binding NACHT domain of NLRs and explore recent breakthroughs that generate connections between NLR protein structure and function. Indeed, newly deposited NLR structures for NLRC4 and NLRP3 have provided unique perspectives on the ATP-dependency of inflammasome activation. Novel molecular dynamic simulations of NLRP3 examined the active site of ADP- and ATP-bound models. The findings support distinctions in nucleotide-binding domain topology with occupancy of ATP or ADP that are in turn disseminated on to the global protein structure. -

Supplemental Information
Supplemental information Dissection of the genomic structure of the miR-183/96/182 gene. Previously, we showed that the miR-183/96/182 cluster is an intergenic miRNA cluster, located in a ~60-kb interval between the genes encoding nuclear respiratory factor-1 (Nrf1) and ubiquitin-conjugating enzyme E2H (Ube2h) on mouse chr6qA3.3 (1). To start to uncover the genomic structure of the miR- 183/96/182 gene, we first studied genomic features around miR-183/96/182 in the UCSC genome browser (http://genome.UCSC.edu/), and identified two CpG islands 3.4-6.5 kb 5’ of pre-miR-183, the most 5’ miRNA of the cluster (Fig. 1A; Fig. S1 and Seq. S1). A cDNA clone, AK044220, located at 3.2-4.6 kb 5’ to pre-miR-183, encompasses the second CpG island (Fig. 1A; Fig. S1). We hypothesized that this cDNA clone was derived from 5’ exon(s) of the primary transcript of the miR-183/96/182 gene, as CpG islands are often associated with promoters (2). Supporting this hypothesis, multiple expressed sequences detected by gene-trap clones, including clone D016D06 (3, 4), were co-localized with the cDNA clone AK044220 (Fig. 1A; Fig. S1). Clone D016D06, deposited by the German GeneTrap Consortium (GGTC) (http://tikus.gsf.de) (3, 4), was derived from insertion of a retroviral construct, rFlpROSAβgeo in 129S2 ES cells (Fig. 1A and C). The rFlpROSAβgeo construct carries a promoterless reporter gene, the β−geo cassette - an in-frame fusion of the β-galactosidase and neomycin resistance (Neor) gene (5), with a splicing acceptor (SA) immediately upstream, and a polyA signal downstream of the β−geo cassette (Fig. -

Epigenome-Wide Study Identified Methylation Sites Associated With
nutrients Article Epigenome-Wide Study Identified Methylation Sites Associated with the Risk of Obesity Majid Nikpay 1,*, Sepehr Ravati 2, Robert Dent 3 and Ruth McPherson 1,4,* 1 Ruddy Canadian Cardiovascular Genetics Centre, University of Ottawa Heart Institute, 40 Ruskin St–H4208, Ottawa, ON K1Y 4W7, Canada 2 Plastenor Technologies Company, Montreal, QC H2P 2G4, Canada; [email protected] 3 Department of Medicine, Division of Endocrinology, University of Ottawa, the Ottawa Hospital, Ottawa, ON K1Y 4E9, Canada; [email protected] 4 Atherogenomics Laboratory, University of Ottawa Heart Institute, Ottawa, ON K1Y 4W7, Canada * Correspondence: [email protected] (M.N.); [email protected] (R.M.) Abstract: Here, we performed a genome-wide search for methylation sites that contribute to the risk of obesity. We integrated methylation quantitative trait locus (mQTL) data with BMI GWAS information through a SNP-based multiomics approach to identify genomic regions where mQTLs for a methylation site co-localize with obesity risk SNPs. We then tested whether the identified site contributed to BMI through Mendelian randomization. We identified multiple methylation sites causally contributing to the risk of obesity. We validated these findings through a replication stage. By integrating expression quantitative trait locus (eQTL) data, we noted that lower methylation at cg21178254 site upstream of CCNL1 contributes to obesity by increasing the expression of this gene. Higher methylation at cg02814054 increases the risk of obesity by lowering the expression of MAST3, whereas lower methylation at cg06028605 contributes to obesity by decreasing the expression of SLC5A11. Finally, we noted that rare variants within 2p23.3 impact obesity by making the cg01884057 Citation: Nikpay, M.; Ravati, S.; Dent, R.; McPherson, R. -

Coincidental Loss of DOCK8 Function in NLRP10-Deficient and C3H/Hej Mice Results in Defective Dendritic Cell Migration
Coincidental loss of DOCK8 function in NLRP10-deficient and C3H/HeJ mice results in defective dendritic cell migration Jayendra Kumar Krishnaswamya,b,1, Arpita Singha,b,1, Uthaman Gowthamana,b, Renee Wua,b, Pavane Gorrepatia,b, Manuela Sales Nascimentoa,b, Antonia Gallmana,b, Dong Liua,b, Anne Marie Rhebergenb, Samuele Calabroa,b, Lan Xua,b, Patricia Ranneya,b, Anuj Srivastavac, Matthew Ransond, James D. Gorhamd, Zachary McCawe, Steven R. Kleebergere, Leonhard X. Heinzf, André C. Müllerf, Keiryn L. Bennettf, Giulio Superti-Furgaf, Jorge Henao-Mejiag, Fayyaz S. Sutterwalah, Adam Williamsi, Richard A. Flavellb,j,2, and Stephanie C. Eisenbartha,b,2 Departments of aLaboratory Medicine and bImmunobiology and jHoward Hughes Medical Institute, Yale University School of Medicine, New Haven, CT 06520; cComputational Sciences, The Jackson Laboratory, Bar Harbor, ME 04609; dDepartment of Pathology, Geisel School of Medicine at Dartmouth, Hanover, NH 03755; eNational Institute of Environmental Health Sciences, Research Triangle Park, NC 27709; fCeMM Research Center for Molecular Medicine of the Austrian Academy of Sciences, 1090 Vienna, Austria; gInstitute for Immunology, Departments of Pathology and Laboratory Medicine, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA 19104; hInflammation Program, Department of Internal Medicine, University of Iowa, Iowa City, IA 52241; and iThe Jackson Laboratory for Genomic Medicine, Department of Genetics and Genome Sciences, University of Connecticut Health Center, Farmington, CT 06032 Contributed by Richard A. Flavell, January 28, 2015 (sent for review December 23, 2014; reviewed by Matthew L. Albert and Thirumala-Devi Kanneganti) Dendritic cells (DCs) are the primary leukocytes responsible for NLRP10 is the only NOD-like receptor (NLR) without a leu- priming T cells. -

Characterization of Its Binding to Hematopoietic Cell Kinase Yue Zhang and Curtis T
Int. J. Biol. Sci. 2020, Vol. 16 1507 Ivyspring International Publisher International Journal of Biological Sciences 2020; 16(9): 1507-1525. doi: 10.7150/ijbs.41798 Research Paper Nucleotide binding domain and leucine-rich repeat pyrin domain-containing protein 12: characterization of its binding to hematopoietic cell kinase Yue Zhang and Curtis T. Okamoto Department of Pharmacology and Pharmaceutical Sciences, School of Pharmacy, University of Southern California, USA 90089-9121 Corresponding author: Yue Zhang, [email protected] © The author(s). This is an open access article distributed under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0/). See http://ivyspring.com/terms for full terms and conditions. Received: 2019.11.04; Accepted: 2020.02.13; Published: 2020.03.05 Abstract Protein-protein interactions are key to define the function of nucleotide binding domain (NBD) and leucine-rich repeat (LRR) family, pyrin domain (PYD)-containing protein 12 (NLRP12). cDNA encoding the human PYD + NBD of NLRP12 was used as bait in a yeast two-hybrid screen with a human leukocyte cDNA library as prey. Hematopoiesis cell kinase (HCK), a member of the c-SRC family of non-receptor tyrosine kinases, was among the top hits. The C-terminal 40 amino acids of HCK selectively bound to NLRP12’s PYD + NBD, but not to that of NLRP3 and NLRP8. Amino acids F503, I506, Q507, L510, and D511 of HCK are critical for the binding of HCK’s C-terminal 40 amino acids to NLRP12’s PYD + NBD. Additionally, the C-terminal 30 amino acids of HCK are sufficient to bind to NLRP12’s PYD + NBD, but not to its PYD alone nor to its NBD alone. -

NLR Members in Inflammation-Associated
Cellular & Molecular Immunology (2017) 14, 403–405 & 2017 CSI and USTC All rights reserved 2042-0226/17 $32.00 www.nature.com/cmi RESEARCH HIGHTLIGHT NLR members in inflammation-associated carcinogenesis Ha Zhu1,2 and Xuetao Cao1,2,3 Cellular & Molecular Immunology (2017) 14, 403–405; doi:10.1038/cmi.2017.14; published online 3 April 2017 hronic inflammation is regarded as an impor- nucleotide-binding and oligomerization domain IL-2,8 and NAIP was found to regulate the STAT3 Ctant factor in cancer progression. In addition (NOD)-like receptors (NLRs). TLRs and CLRs are pathway independent of inflammasome formation.9 to the immune surveillance function in the early located in the plasma membranes, whereas RLRs, The AOM/DSS model is the most popular model stage of tumorigenesis, inflammation is also known ALRs and NLRs are intracellular PRRs.3 Unlike used to study the function of NLRs in fl fl as one of the hallmarks of cancer and can supply other families that have been shown to bind their in ammation-associated carcinogenesis. In amma- the tumor microenvironment with bioactive mole- specific cognate ligands, the distinct ligands for somes initiated by NLRs or AIM2 have been widely cules and favor the development of other hallmarks NLRs are still unknown. In fact, mounting evidence reported to participate in the maintenance of 10,11 Nlrp3 Nlrp6 of cancer, such as genetic instability and angiogen- suggests that NLRs function as cytoplasmic sensors intestinal homeostasis. -/-, -/-, Nlrc4 Nlrp1 Nlrx1 Nlrp12 esis. Moreover, inflammation contributes to the and participate in modulating TLR, RLR and CLR -/-, -/-, -/- and -/- mice are 4 more susceptible to AOM/DSS-induced colorectal changing tumor microenvironment by altering signaling pathways.