Chapter 1 Ozone-Depleting Substances (Odss) and Related Chemicals
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Destabilizing Thermohaline Circulation–Atmosphere–Sea Ice
642 JOURNAL OF CLIMATE VOLUME 12 NOTES AND CORRESPONDENCE A Destabilizing Thermohaline Circulation±Atmosphere±Sea Ice Feedback STEVEN R. JAYNE MIT±WHOI Joint Program in Oceanography, Woods Hole Oceanographic Institution, Woods Hole, Massachusetts JOCHEM MAROTZKE Center for Global Change Science, Department of Earth, Atmospheric and Planetary Sciences, Massachusetts Institute of Technology, Cambridge, Massachusetts 18 November 1996 and 9 March 1998 ABSTRACT Some of the interactions and feedbacks between the atmosphere, thermohaline circulation, and sea ice are illustrated using a simple process model. A simpli®ed version of the annual-mean coupled ocean±atmosphere box model of Nakamura, Stone, and Marotzke is modi®ed to include a parameterization of sea ice. The model includes the thermodynamic effects of sea ice and allows for variable coverage. It is found that the addition of sea ice introduces feedbacks that have a destabilizing in¯uence on the thermohaline circulation: Sea ice insulates the ocean from the atmosphere, creating colder air temperatures at high latitudes, which cause larger atmospheric eddy heat and moisture transports and weaker oceanic heat transports. These in turn lead to thicker ice coverage and hence establish a positive feedback. The results indicate that generally in colder climates, the presence of sea ice may lead to a signi®cant destabilization of the thermohaline circulation. Brine rejection by sea ice plays no important role in this model's dynamics. The net destabilizing effect of sea ice in this model is the result of two positive feedbacks and one negative feedback and is shown to be model dependent. To date, the destabilizing feedback between atmospheric and oceanic heat ¯uxes, mediated by sea ice, has largely been neglected in conceptual studies of thermohaline circulation stability, but it warrants further investigation in more realistic models. -

Tropical Climate
UGAMP: A network of excellence in climate modelling and research Issue 27 October 2003 UGAMP Coordinator: Prof. Julia Slingo [email protected] Newsletter Editor: Dr. Glenn Carver [email protected] Newsletter website: acmsu.nerc.ac.uk/newsletter.html Contents NCAS News . 2 NCAS Websites . 3 NCAS Centres and Facilities . 3 UGAMP Coordinator . 4 CGAM Director . 4 ACMSU Director . 4 HPC Facilities . 5 New areas of UGAMP science 7 Chemistry-climate interactions . 19 Climate variability and predictability . 32 Atmospheric Composition . 48 Tropospheric chemistry and aerosols . 58 Climate Dynamics . 64 Model development . 72 Group News . 78 (for full contents see listing on the inside back cover) NERC Centres for Atmospheric Science, NCAS Alan Thorpe ([email protected]): Director NCAS Since the last UGAMP Newsletter there have been a significant number of NCAS developments relevant to the UK atmospheric science community. These include the following, which are particularly pertinent to the UGAMP community: • NERC have agreed to fund a new directed (new name for thematic) programme called “Surface Ocean – Lower Atmosphere Study” or SOLAS for short. • NERC have agreed to fund a “pump-priming” activity for a proposed new directed programme called Flood Risk from Extreme Events, FREE. The full proposal for FREE will be considered by NERC early in 2004. •NCAS is supporting a project to develop a new chemistry module for the HadGEM model. This is called UK-CHEM and Olaf Morgenstern at ACMSU is collaborating closely with the Hadley Centre on the project. •NCAS is supporting a project to develop the science for a new aerosol module for HadGEM. -

Results from the Implementation of the Elastic Viscous Plastic Sea Ice Rheology in Hadcm3 W
Results from the implementation of the Elastic Viscous Plastic sea ice rheology in HadCM3 W. M. Connolley, A. B. Keen, A. J. Mclaren To cite this version: W. M. Connolley, A. B. Keen, A. J. Mclaren. Results from the implementation of the Elastic Viscous Plastic sea ice rheology in HadCM3. Ocean Science, European Geosciences Union, 2006, 2 (2), pp.201- 211. hal-00298295 HAL Id: hal-00298295 https://hal.archives-ouvertes.fr/hal-00298295 Submitted on 23 Oct 2006 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Ocean Sci., 2, 201–211, 2006 www.ocean-sci.net/2/201/2006/ Ocean Science © Author(s) 2006. This work is licensed under a Creative Commons License. Results from the implementation of the Elastic Viscous Plastic sea ice rheology in HadCM3 W. M. Connolley1, A. B. Keen2, and A. J. McLaren2 1British Antarctic Survey, High Cross, Madingley Road, Cambridge, CB3 0ET, UK 2Met Office Hadley Centre, FitzRoy Road, Exeter, EX1 3PB, UK Received: 13 June 2006 – Published in Ocean Sci. Discuss.: 10 July 2006 Revised: 21 September 2006 – Accepted: 16 October 2006 – Published: 23 October 2006 Abstract. We present results of an implementation of the a full dynamical model incorporating wind stresses and in- Elastic Viscous Plastic (EVP) sea ice dynamics scheme into ternal ice stresses leads to errors in the detailed representa- the Hadley Centre coupled ocean-atmosphere climate model tion of sea ice and limits our confidence in its future predic- HadCM3. -
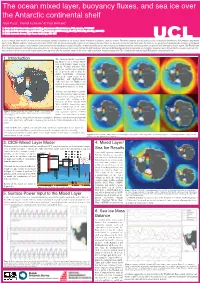
3. CICE-Mixed Layer Model 4. Mixed Layer/ Sea Ice Results 5. Surface
The ocean mixed layer, buoyancy fluxes, and sea ice over the Antarctic continental shelf Alek Petty1, Daniel Feltham2 & Paul Holland3 1. Centre for Polar Observation and Modelling, Department of Earth Sciences, UCL, London, WC1E6BT 2. Centre for Polar Observation and Modelling, Department of Meteorology, Reading University, Reading, RG6 6BB 2. British Antarctic Survey, High Cross, Cambridge, CB3 0ET A sea ice-mixed layer model has been used to investigate regional variations in the surface-driven formation of Antarctic shelf sea waters. The model captures well the expected sea ice thickness distribution, and produces deep mixed layers in the Weddell and Ross shelf seas each winter (1985-2011). By deconstructing the surface power input to the mixed layer, we have shown that the salt/fresh water flux from sea ice growth/melt dominates the evolution of the mixed layer in all shelf sea regions, with a smaller contribution from the mixed layer-surface heat flux. An analysis of the sea ice mass balance has demonstrated the contrasting mean ice growth, melt and export in each region. The Weddell and Ross shelf seas expereince the highest annual ice growth, with a large fraction of this ice exported northwards each year, whereas the Bellingshausen shelf sea experiences the highest annual ice melt, despite the low annual ice growth. Cur- rent work (not shown) is focussed on atmospheric forcing trends and the resultant trends in the sea ice and mixed layer evolution using both ERA-I hindcast forcing and hadGEM2 future climate projections. 1. Introduction The continental shelf seas surround- ing Antarctica are a crucial compo- nent of the Earth’s climate system, with the Weddell and Ross (WR) shelf seas cooling and ventilating the deep ocean and feeding the global thermohaline circulation, whereas the warm waters in the Amundsen and Bellingshausen (AB) shelf seas (see Figure 1) are implicit in the recent ocean-driven melting of the Antarctic ice sheet. -

Test Plan: CICE Sea Ice Model for NGGPS
Test Plan: CICE Sea Ice Model for NGGPS Final 16 March 2016 POC: Shan Sun – NOAA ESRL/GSD/GMTB – [email protected] Introduction After reviewing several sea ice models at the Sea Ice Workshop organized by Global Modeling Test Bed (GMTB) in February 2016, the committee has selected the Los Alamos Community Ice CodE (CICE) as the sea ice model to be incorporated as a component of Next-Generation Global Prediction System (NGGPS). The GMTB is proposing to carry out testing and evaluations with this model in two phases as the next step. Testing framework Hebert et al. (2015) showed that the Arctic Cap Nowcast/Forecast System (ACNFS) demonstrated a high level of skill compared to persistence in 1-7 day forecasts over a period of one year. ACNFS uses CICE version 4.0 [Hunke and Lipscomb, 2008] as the sea ice model two-way coupled to the HYbrid Coordinate Ocean Model (HYCOM) [Bleck, 2002; Metzger et al., 2014, 2015]. Building on this study, we propose to base the test on a similar configuration. In order to make the test most relevant for NGGPS, there will be three important differences with respect to the Herbert et al. (2015) configuration. First, CICE will be upgraded to its latest version v5, [in which both the code structure and the state variables are similar to v4. The CICE5 code does include a number of new physics options such as the mushy-layer thermodynamics and two new melt pond parameterizations.] V5.1.2 is available at http://oceans11.lanl.gov/svn/CICE/tags/release-5.1.2. -

A New Dynamical Downscaling Approach with GCM Bias
PUBLICATIONS Journal of Geophysical Research: Atmospheres RESEARCH ARTICLE A new dynamical downscaling approach with GCM 10.1002/2014JD022958 bias corrections and spectral nudging Key Points: Zhongfeng Xu1 and Zong-Liang Yang2,3 • Both GCM and RCM biases should be constrained in regional climate 1RCE-TEA and Young Scientist Laboratory, Institute of Atmospheric Physics, Chinese Academy of Sciences, Beijing, China, projection 2 3 • The NDD approach significantly RCE-TEA, Institute of Atmospheric Physics, Chinese Academy of Sciences, Beijing, China, Department of Geological improves the downscaled climate Sciences, Jackson School of Geosciences, University of Texas at Austin, Austin, Texas, USA • NDD is designed for regional climate projection at various time scales Abstract To improve confidence in regional projections of future climate, a new dynamical downscaling Supporting Information: (NDD) approach with both general circulation model (GCM) bias corrections and spectral nudging is developed • Figures S1–S4 and assessed over North America. GCM biases are corrected by adjusting GCM climatological means and • Figure S1 variances based on reanalysis data before the GCM output is used to drive a regional climate model (RCM). • Figure S2 • Figure S3 Spectral nudging is also applied to constrain RCM-based biases. Three sets of RCM experiments are integrated • Figure S4 over a 31 year period. In the first set of experiments, the model configurations are identical except that the initial and lateral boundary conditions are derived from either the original GCM output, the bias-corrected GCM output, Correspondence to: orthereanalysisdata.Thesecondset of experiments is the same as the first set except spectral nudging is applied. Z.-L. Yang, [email protected] The third set of experiments includes two sensitivity runs with both GCM bias corrections and nudging where the nudging strength is progressively reduced. -

Assimila Blank
NERC NERC Strategy for Earth System Modelling: Technical Support Audit Report Version 1.1 December 2009 Contact Details Dr Zofia Stott Assimila Ltd 1 Earley Gate The University of Reading Reading, RG6 6AT Tel: +44 (0)118 966 0554 Mobile: +44 (0)7932 565822 email: [email protected] NERC STRATEGY FOR ESM – AUDIT REPORT VERSION1.1, DECEMBER 2009 Contents 1. BACKGROUND ....................................................................................................................... 4 1.1 Introduction .............................................................................................................. 4 1.2 Context .................................................................................................................... 4 1.3 Scope of the ESM audit ............................................................................................ 4 1.4 Methodology ............................................................................................................ 5 2. Scene setting ........................................................................................................................... 7 2.1 NERC Strategy......................................................................................................... 7 2.2 Definition of Earth system modelling ........................................................................ 8 2.3 Broad categories of activities supported by NERC ................................................. 10 2.4 Structure of the report ........................................................................................... -

Review of the Global Models Used Within Phase 1 of the Chemistry–Climate Model Initiative (CCMI)
Geosci. Model Dev., 10, 639–671, 2017 www.geosci-model-dev.net/10/639/2017/ doi:10.5194/gmd-10-639-2017 © Author(s) 2017. CC Attribution 3.0 License. Review of the global models used within phase 1 of the Chemistry–Climate Model Initiative (CCMI) Olaf Morgenstern1, Michaela I. Hegglin2, Eugene Rozanov18,5, Fiona M. O’Connor14, N. Luke Abraham17,20, Hideharu Akiyoshi8, Alexander T. Archibald17,20, Slimane Bekki21, Neal Butchart14, Martyn P. Chipperfield16, Makoto Deushi15, Sandip S. Dhomse16, Rolando R. Garcia7, Steven C. Hardiman14, Larry W. Horowitz13, Patrick Jöckel10, Beatrice Josse9, Douglas Kinnison7, Meiyun Lin13,23, Eva Mancini3, Michael E. Manyin12,22, Marion Marchand21, Virginie Marécal9, Martine Michou9, Luke D. Oman12, Giovanni Pitari3, David A. Plummer4, Laura E. Revell5,6, David Saint-Martin9, Robyn Schofield11, Andrea Stenke5, Kane Stone11,a, Kengo Sudo19, Taichu Y. Tanaka15, Simone Tilmes7, Yousuke Yamashita8,b, Kohei Yoshida15, and Guang Zeng1 1National Institute of Water and Atmospheric Research (NIWA), Wellington, New Zealand 2Department of Meteorology, University of Reading, Reading, UK 3Department of Physical and Chemical Sciences, Universitá dell’Aquila, L’Aquila, Italy 4Environment and Climate Change Canada, Montréal, Canada 5Institute for Atmospheric and Climate Science, ETH Zürich (ETHZ), Zürich, Switzerland 6Bodeker Scientific, Christchurch, New Zealand 7National Center for Atmospheric Research (NCAR), Boulder, Colorado, USA 8National Institute of Environmental Studies (NIES), Tsukuba, Japan 9CNRM UMR 3589, Météo-France/CNRS, -
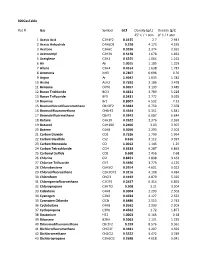
Gas Conversion Factor for 300 Series
300GasTable Rec # Gas Symbol GCF Density (g/L) Density (g/L) 25° C / 1 atm 0° C / 1 atm 1 Acetic Acid C2H4F2 0.4155 2.7 2.947 2 Acetic Anhydride C4H6O3 0.258 4.173 4.555 3 Acetone C3H6O 0.3556 2.374 2.591 4 Acetonitryl C2H3N 0.5178 1.678 1.832 5 Acetylene C2H2 0.6255 1.064 1.162 6 Air Air 1.0015 1.185 1.293 7 Allene C3H4 0.4514 1.638 1.787 8 Ammonia NH3 0.7807 0.696 0.76 9 Argon Ar 1.4047 1.633 1.782 10 Arsine AsH3 0.7592 3.186 3.478 11 Benzene C6H6 0.3057 3.193 3.485 12 Boron Trichloride BCl3 0.4421 4.789 5.228 13 Boron Triflouride BF3 0.5431 2.772 3.025 14 Bromine Br2 0.8007 6.532 7.13 15 Bromochlorodifluoromethane CBrClF2 0.3684 6.759 7.378 16 Bromodifluoromethane CHBrF2 0.4644 5.351 5.841 17 Bromotrifluormethane CBrF3 0.3943 6.087 6.644 18 Butane C4H10 0.2622 2.376 2.593 19 Butanol C4H10O 0.2406 3.03 3.307 20 Butene C4H8 0.3056 2.293 2.503 21 Carbon Dioxide CO2 0.7526 1.799 1.964 22 Carbon Disulfide CS2 0.616 3.112 3.397 23 Carbon Monoxide CO 1.0012 1.145 1.25 24 Carbon Tetrachloride CCl4 0.3333 6.287 6.863 25 Carbonyl Sulfide COS 0.668 2.456 2.68 26 Chlorine Cl2 0.8451 2.898 3.163 27 Chlorine Trifluoride ClF3 0.4496 3.779 4.125 28 Chlorobenzene C6H5Cl 0.2614 4.601 5.022 29 Chlorodifluoroethane C2H3ClF2 0.3216 4.108 4.484 30 Chloroform CHCl3 0.4192 4.879 5.326 31 Chloropentafluoroethane C2ClF5 0.2437 6.314 6.892 32 Chloropropane C3H7Cl 0.308 3.21 3.504 33 Cisbutene C4H8 0.3004 2.293 2.503 34 Cyanogen C2N2 0.4924 2.127 2.322 35 Cyanogen Chloride ClCN 0.6486 2.513 2.743 36 Cyclobutane C4H8 0.3562 2.293 2.503 37 Cyclopropane C3H6 0.4562 -

NWP • 2008 Supercomputer Procurement Status
FromFrom ClimateClimate toto WeatherWeather CoupledCoupled Models,Models, ChallengesChallenges forfor ACCESSACCESS www.cawcr.gov.au Tim F. Pugh Thirteenth Workshop on the Use of High Performance Computing in Meteorology 3 - 7 November 2008 The Centre for Australian Weather and Climate Research A partnership between CSIRO and the Bureau of Meteorology OutlineOutline ofof TalkTalk • Introduction to the Australian Community Climate Earth-System Simulator (ACCESS) program initiated in 2006 • … and the Centre for Australian Weather and Climate Research (CAWCR) initiated in 2007 • ACCESS status today • Earth System Modelling for Climate • Numerical Weather Prediction • Numerical Ocean Prediction • ACCESS directions and challenges • Earth System Modelling for Climate • Earth System Modelling for NWP • 2008 supercomputer procurement status The Centre for Australian Weather and Climate Research A partnership between CSIRO and the Bureau of Meteorology IntroductionIntroduction toto … … ACCESS The Australian Community Climate Earth-System Simulator ACCESS Objectives are to… • Develop a national approach to climate change and weather prediction system development and research, particularly as it manifests in the Australian region • Focus on the needs of a wide range of stakeholders: • Providing the best possible services • Analysing climate impacts and adaptation • Linkages with relevant University research • Meeting policy needs in natural resource management ACCESS is a joint initiative of the Bureau of Meteorology and CSIRO in cooperation with the university community in Australia http://www.accessimulator.org.au/ The Centre for Australian Weather and Climate Research A partnership between CSIRO and the Bureau of Meteorology IntroductionIntroduction toto CAWCRCAWCR QuickTime™ and a QuickTime™ and a TIFF (Uncompressed) decompressor The Centre for Australian Weather and Climate Research TIFF (Uncompressed) decompressor are needed to see this picture. -

Recent and Future Development of the US Navy Numerical Weather Prediction Systems
Recent and Future Development of the US Navy Numerical Weather Prediction Systems Melinda Peng Marine Meteorology Division Naval Research Laboratory Monterey, CA 93943 Abstract The Naval Research Laboratory (NRL) is responsible for developing, transitioning, and maintaining the Navy’s numerical weather prediction systems that provide a unique environmental prediction capability demanded by the warfighters. The current NRL modeling suites consist of 1) the NAVy Global Environmental Model (NAVGEM); 2) the Coupled Ocean/Atmosphere Mesoscale Prediction System (COAMPS®); 3) NRL Atmospheric Variational Data Assimilation System–Accelerated Representer (NAVDAS-AR), an observation space 4DVar for NAVGEM , and NRL Atmospheric Variational Data Assimilation System (NAVDAS), a 3DVar for COAMPS, respectively. NRL recently added a hybrid data assimilation capability to include the flow-dependent background error information for global data assimilation. The COAMPS 4DVar has been developed to replace the COAMPS 3DVar for mesoscale data assimilation. COAMPS has been coupled in a two-way interactive sense to the NRL Coastal Ocean Model (NCOM) and the SWAN and WWIII wave models using the Earth System Modeling Framework (ESMF). Additional capabilities under development include a fully coupled sea ice model (Los Alamos CICE) capability and an integrated hydrological modeling component for high-resolution COAMPS coastal applications. The tropical cyclone system, COAMPS-TC, was transitioned to Navy operations in 2013, one of the top performing dynamical models worldwide for tropical cyclone intensity prediction. In 2016, a fully coupled version of COAMPS-TC coupled with NCOM has been implemented in operations. NRL is developing a fully coupled global extended-range prediction system including the Navy Global Environmental Model (NAVGEM), the HYbrid Coordinate Ocean Model (HYCOM), the Los Alamos Sea Ice Model (CICE), and the Wavewatch III ocean surface wave model as part of the national Earth System Prediction Capability (ESPC). -

(12) United States Patent (10) Patent No.: US 8,911,640 B2 Nappa Et Al
USOO891. 1640B2 (12) United States Patent (10) Patent No.: US 8,911,640 B2 Nappa et al. (45) Date of Patent: Dec. 16, 2014 (54) COMPOSITIONS COMPRISING 5,736,063 A 4/1998 Richard et al. FLUOROOLEFNS AND USES THEREOF 5,744,052 A 4/1998 Bivens 5,788,886 A 8, 1998 Minor et al. 5,897.299 A * 4/1999 Fukunaga ..................... 417.316 (71) Applicant: E I du Pont de Nemours and 5,969,198 A 10/1999 Thenappan et al. Company, Wilmington, DE (US) 6,053,008 A 4/2000 Arman et al. 6,065.305 A 5/2000 Arman et al. (72) Inventors: Mario Joseph Nappa, Newark, DE 6,076,372 A 6/2000 Acharya et al. 6,111,150 A 8/2000 Sakyu et al. (US); Barbara Haviland Minor, Elkton, 6,176,102 B1 1/2001 Novak et al. MD (US); Allen Capron Sievert, 6,258,292 B1 7/2001 Turner Elkton, MD (US) 6,300,378 B1 10/2001 Tapscott 6.426,019 B1 7/2002 Acharya et al. (73) Assignee: E I du Pont de Nemours and 6,610,250 B1 8, 2003 Tuma Company, Wilmington, DE (US) 6,858,571 B2 2/2005 Pham et al. 6,969,701 B2 11/2005 Singh et al. 7,708,903 B2 5, 2010 Sievert et al. *) Notice: Subject to anyy disclaimer, the term of this 8,012,368 B2 9/2011 Nappa et al. patent is extended or adjusted under 35 8,070,976 B2 12/2011 Nappa et al. U.S.C. 154(b) by 0 days.