Factor XII Deficiency Mimicking Bleeding Diathesis: a Unique Presentation and Diagnostic Pitfall
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Guideline on Clinical Investigation of Recombinant and Human Plasma-Derived 9 Factor IX Products’ (EMA/CHMP/BPWP/144552/2009 Rev
1 15 November 2018 2 EMA/CHMP/BPWP/144552/2009 rev. 2 Corr. 1 3 Committee for medicinal products for human use (CHMP) 4 Guideline on clinical investigation of recombinant and 5 human plasma-derived factor IX products 6 Draft Draft Agreed by Blood Products Working Party (BPWP) August 2018 Adopted by Committee for Medicinal Products for Human Use (CHMP) 15 November 2018 Start of public consultation 3 December 2018 End of public consultation 30 June 2019 7 8 This guideline replaces ‘Guideline on clinical investigation of recombinant and human plasma-derived 9 factor IX products’ (EMA/CHMP/BPWP/144552/2009 Rev. 1, Corr. 1) 10 Comments should be provided using this template. The completed comments form should be sent to [email protected] 11 Keywords Recombinant factor IX, plasma-derived factor IX, efficacy, safety, immunogenicity, inhibitor, thrombogenicity, anaphylactic reactions, potency assays 30 Churchill Place ● Canary Wharf ● London E14 5EU ● United Kingdom Telephone +44 (0)20 3660 6000 Facsimile +44 (0)20 3660 5555 Send a question via our website www.ema.europa.eu/contact An agency of the European Union © European Medicines Agency, 2019. Reproduction is authorised provided the source is acknowledged. 12 Guideline on the clinical investigation of recombinant and 13 human plasma-derived factor IX products 14 Table of contents 15 Executive summary ..................................................................................... 4 16 1. Introduction (background) ..................................................................... -

MONONINE (“Difficulty ® Monoclonal Antibody Purified in Concentrating”; Subject Recovered)
CSL Behring IU/kg (n=38), 0.98 ± 0.45 K at doses >95-115 IU/kg (n=21), 0.70 ± 0.38 K at doses >115-135 IU/kg (n=2), 0.67 K at doses >135-155 IU/kg (n=1), and 0.73 ± 0.34 K at doses >155 IU/kg (n=5). Among the 36 subjects who received these high doses, only one (2.8%) Coagulation Factor IX (Human) reported an adverse experience with a possible relationship to MONONINE (“difficulty ® Monoclonal Antibody Purified in concentrating”; subject recovered). In no subjects were thrombo genic complications MONONINE observed or reported.4 only The manufacturing procedure for MONONINE includes multiple processing steps that DESCRIPTION have been designed to reduce the risk of virus transmission. Validation studies of the Coagulation Factor IX (Human), MONONINE® is a sterile, stable, lyophilized concentrate monoclonal antibody (MAb) immunoaffinity chromatography/chemical treatment step and of Factor IX prepared from pooled human plasma and is intended for use in therapy nanofiltration step used in the production of MONONINE doc ument the virus reduction of Factor IX deficiency, known as Hemophilia B or Christmas disease. MONONINE is capacity of the processes employed. These studies were conducted using the rel evant purified of extraneous plasma-derived proteins, including Factors II, VII and X, by use of enveloped and non-enveloped viruses. The results of these virus validation studies utilizing immunoaffinity chromatography. A murine monoclonal antibody to Factor IX is used as an a wide range of viruses with different physicochemical properties are summarized in Table affinity ligand to isolate Factor IX from the source material. -

Section 8: Hematology CHAPTER 47: ANEMIA
Section 8: Hematology CHAPTER 47: ANEMIA Q.1. A 56-year-old man presents with symptoms of severe dyspnea on exertion and fatigue. His laboratory values are as follows: Hemoglobin 6.0 g/dL (normal: 12–15 g/dL) Hematocrit 18% (normal: 36%–46%) RBC count 2 million/L (normal: 4–5.2 million/L) Reticulocyte count 3% (normal: 0.5%–1.5%) Which of the following caused this man’s anemia? A. Decreased red cell production B. Increased red cell destruction C. Acute blood loss (hemorrhage) D. There is insufficient information to make a determination Answer: A. This man presents with anemia and an elevated reticulocyte count which seems to suggest a hemolytic process. His reticulocyte count, however, has not been corrected for the degree of anemia he displays. This can be done by calculating his corrected reticulocyte count ([3% × (18%/45%)] = 1.2%), which is less than 2 and thus suggestive of a hypoproliferative process (decreased red cell production). Q.2. A 25-year-old man with pancytopenia undergoes bone marrow aspiration and biopsy, which reveals profound hypocellularity and virtual absence of hematopoietic cells. Cytogenetic analysis of the bone marrow does not reveal any abnormalities. Despite red blood cell and platelet transfusions, his pancytopenia worsens. Histocompatibility testing of his only sister fails to reveal a match. What would be the most appropriate course of therapy? A. Antithymocyte globulin, cyclosporine, and prednisone B. Prednisone alone C. Supportive therapy with chronic blood and platelet transfusions only D. Methotrexate and prednisone E. Bone marrow transplant Answer: A. Although supportive care with transfusions is necessary for treating this patient with aplastic anemia, most cases are not self-limited. -

Method Development and Validation of Vitamin D2 and Vitamin D3 Using Mass Spectrometry
Method Development and Validation of Vitamin D2 and Vitamin D3 Using Mass Spectrometry Devon Victoria Riley A thesis submitted in partial fulfillment of the requirements for the degree of Master of Science University of Washington 2016 Committee: Andrew Hoofnagle Geoffrey Baird Dina Greene Program Authorized to Offer Degree: Laboratory Medicine ©Copyright 2016 Devon V. Riley ii University of Washington Abstract Method Development and Validation of Vitamin D2 and Vitamin D3 Using Mass Spectrometry Devon V. Riley Chair of the Supervisory Committee: Associate Professor Andrew Hoofnagle, MD, PhD Vitamin D has long been known to maintain bone health by regulating calcium and phosphorous homeostasis. In recent years, scientists have discovered additional physiological roles for vitamin D. The complex interaction between the active vitamin D hormone and its metabolic precursors continues to be a rich area of research. Fundamental to this research is the availability of accurate and precise assays. Few published assays for vitamins D2 and D3 have contained sufficient details on method validation or performance characteristics. The liquid chromatography-tandem mass spectrometry (LC-MS/MS) assay developed for this thesis has undergone a rigorous validation and proven to yield a sensitive and specific method that exceeds the capabilities of all previously published methods. Developing and validating a novel assay is often complicated by the lack of established acceptability standards. This thesis explores this challenge, specifically for establishing meaningful interpretations and qualification standards of the lower limit of the measuring interval. Altogether, future research focused on vitamins D2, D3 and the Vitamin D pathway can benefit from this robust LC-MS/MS assay and the associated quality parameters outlined in this thesis. -

Familial Multiple Coagulation Factor Deficiencies
Journal of Clinical Medicine Article Familial Multiple Coagulation Factor Deficiencies (FMCFDs) in a Large Cohort of Patients—A Single-Center Experience in Genetic Diagnosis Barbara Preisler 1,†, Behnaz Pezeshkpoor 1,† , Atanas Banchev 2 , Ronald Fischer 3, Barbara Zieger 4, Ute Scholz 5, Heiko Rühl 1, Bettina Kemkes-Matthes 6, Ursula Schmitt 7, Antje Redlich 8 , Sule Unal 9 , Hans-Jürgen Laws 10, Martin Olivieri 11 , Johannes Oldenburg 1 and Anna Pavlova 1,* 1 Institute of Experimental Hematology and Transfusion Medicine, University Clinic Bonn, 53127 Bonn, Germany; [email protected] (B.P.); [email protected] (B.P.); [email protected] (H.R.); [email protected] (J.O.) 2 Department of Paediatric Haematology and Oncology, University Hospital “Tzaritza Giovanna—ISUL”, 1527 Sofia, Bulgaria; [email protected] 3 Hemophilia Care Center, SRH Kurpfalzkrankenhaus Heidelberg, 69123 Heidelberg, Germany; ronald.fi[email protected] 4 Department of Pediatrics and Adolescent Medicine, University Medical Center–University of Freiburg, 79106 Freiburg, Germany; [email protected] 5 Center of Hemostasis, MVZ Labor Leipzig, 04289 Leipzig, Germany; [email protected] 6 Hemostasis Center, Justus Liebig University Giessen, 35392 Giessen, Germany; [email protected] 7 Center of Hemostasis Berlin, 10789 Berlin-Schöneberg, Germany; [email protected] 8 Pediatric Oncology Department, Otto von Guericke University Children’s Hospital Magdeburg, 39120 Magdeburg, Germany; [email protected] 9 Division of Pediatric Hematology Ankara, Hacettepe University, 06100 Ankara, Turkey; Citation: Preisler, B.; Pezeshkpoor, [email protected] B.; Banchev, A.; Fischer, R.; Zieger, B.; 10 Department of Pediatric Oncology, Hematology and Clinical Immunology, University of Duesseldorf, Scholz, U.; Rühl, H.; Kemkes-Matthes, 40225 Duesseldorf, Germany; [email protected] B.; Schmitt, U.; Redlich, A.; et al. -

Evaluation of Prolonged Surface Activated Coagulation Time
Biomedical Science Faculty of Health and Society Malm¨oUniversity SE-205 06 Malm¨o Sweden Master programme in Biomedical Surface Science http://edu.mah.se/en/Program/VABSE Evaluation of prolonged surface activated coagulation time Master degree thesis, 30 ECTS Author: Amalie Jesting Supervisors: Sebastian Bj¨orklund,PhD, Lecturer, Malm¨oUniversity Jens Peter Gøtze, MD, DMSc Professor, Chief-Physician, Rigshospitalet Søren Frank Jørgensen, MSc, Senior Lecturer, Metropolitan University College August 2018 MALMO¨ UNIVERSITY Abstract Faculty of Health and Society Master programme in Biomedical Surface Science Evaluation of prolonged surface activated coagulation time by Amalie Jesting Background: Blood coagulation is an essential defense mechanism to prevent bleeding. Disorders in the coagulation system can be severe and blood tests measuring the blood's ability to coagulate are important. Activated partial thromboplastin time (APTT) is a blood test that measures blood coagulation time. An abnormal prolonged APTT can both be associated with a bleeding tendency or a risk of thrombosis. Additional blood tests are needed to discover the cause of a prolonged APTT. One potential test is the APTT mixing study, which can separate samples with and without inhibitors. The aim of this project is to investigate how the cause of a prolonged APTT is evaluated today and to examine if it is possible to indicate the cause of a prolonged APTT using the APTT mixing study performed on routine samples. The goal is to be able to indicate the cause of a prolonged APTT immediately when is it first discovered. This will save time and help guide the physicians in their work with the patient. -

Gene Therapy Expression Vectors Based on the Clotting Factor IX Promoter
Gene Therapy (1999) 6, 1584–1589 1999 Stockton Press All rights reserved 0969-7128/99 $15.00 http://www.stockton-press.co.uk/gt Gene therapy expression vectors based on the clotting Factor IX promoter H Hoag, J Gore, D Barry and CR Mueller Department of Biochemistry and Cancer Research Laboratories, Queen’s University, Kingston, Ontario, Canada The liver is one of the prime targets for gene therapy, and moter. Introduction of this element increases promoter the correction of defects in a variety of clotting factor genes activity at least 20-fold over the proximal promoter alone is one of the main goals of liver-directed therapies. The use when assayed in the human liver cell line Hep G2. This of transcriptional regulatory elements derived from these optimized promoter is significantly more active than the genes may provide for the optimal expression of trans- SV40 enhancer/early promoter. The expression of the opti- duced genes. We have applied our knowledge of the pro- mized Factor IX promoter is also more persistent in the moter structure of the clotting Factor IX gene to design short term. The inclusion of a liver-specific locus control optimized expression vectors for use in gene therapy. The region, derived from the apolipoprotein E/C locus, did not activity of the proximal promoter has been augmented by further augment expression levels. These Factor IX vectors the introduction of a multimerized upstream site which we also exhibit a high degree of tissue specificity, as meas- have previously shown to be a prime regulator of the pro- ured by transfection into breast and muscle cell lines. -

Intracerebral Bleeding in Young Infants Due to Rare Etiologies–A
eona f N tal l o B a io n l r o u g y o J Tewari et al., J Neonatal Biol 2016, 5:2 Journal of Neonatal Biology DOI: 10.4172/2167-0897.1000221 ISSN: 2167-0897 Case Report Open access Intracerebral Bleeding in Young Infants due to Rare Etiologies–A Report of two Cases Vishal Vishnu Tewari1*, Kunal Tewari2 and Ritu Mehta3 1Department of Pediatrics, Army Hospital (Referral and Research), New Delhi, India 2Department of Anaesthesia, Classified Specialist Anesthesia, Base Hospital, New Delhi, India 3Department of Pathology, All India Institute of Medical Sciences, New Delhi, India *Corresponding author: Vishal Vishnu Tewari, Department of Pediatrics, Army Hospital, New Delhi, India,Tel: +91-8826118889, E-mail: [email protected] Rec date: April 29, 2016; Acc date: May 16, 2016; Pub date: May 23, 2016 Copyright: © 2016 Tewari VV, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. Abstract Neonates and young infants are physiologically encumbered by an inadequate hemostatic mechanism. They may also have inherited or acquired bleeding disorders which may have a catastrophic presentation with intracerebral hemorrhage. The need to reach an accurate diagnosis is paramount in order to provide accurate therapy and genetic counseling. We report two infants who presented with unprovoked life threatening massive intracerebral hemorrhage. The first infant was diagnosed and managed for congenital factor V deficiency while the second infant had Glanzmann thrombasthenia. Keywords: Intracerebral bleeding; Congenital factor V deficiency; disorder in the family. -

A Life-Threatening Case of Pregnancy-Related Atypical
Puri et al. BMC Nephrology (2020) 21:488 https://doi.org/10.1186/s12882-020-02100-4 CASE REPORT Open Access A life-threatening case of pregnancy- related atypical Haemolytic uremic syndrome and successful treatment with Eculizumab Prianka Puri1,2* , Anida Hanxhiu1, Daniel V. O’Hara1,3, Danny Hsu4 and Mirna Vucak-Dzumhur1,5 Abstract Background: Pregnancy-related Atypical Haemolytic Uremic Syndrome (P-aHUS) is a rare condition affecting genetically predisposed women during pregnancy. It is often difficult to diagnose and has a significant impact on maternal and foetal outcomes. It is characterised by microangiopathic haemolytic anaemia and kidney injury from thrombotic microangiopathy. Case presentation: A 27-year-old female of Lebanese descent presented at 36 weeks’ gestation with foetal death in-utero (FDIU) with placental abruption on a background of previously normal antenatal visits. She was coagulopathic and anaemic with anuric acute kidney injury, requiring emergency Caesarean section, intubation and dialysis. Her coagulopathy rapidly resolved, however, her anaemia and renal dysfunction persisted. A diagnosis of P-aHUS was made, and she was empirically treated with Eculizumab. Her ADAMTS13 level was normal, effectively excluding thrombotic thrombocytopenic purpura. Within 2 weeks of treatment her haematological parameters improved, and her renal function began to recover and within 2 months she became dialysis independent. Conclusion: This case highlights the challenges of a timely diagnosis of P-aHUS from other pregnancy-related diseases. Although our patient is dialysis-independent, her risk of relapse remains high with subsequent pregnancies. Currently we are awaiting her genetic sequencing to complete her assessment for underlying mutations and are determining the safest approach to a future planned pregnancy. -

Mixing Studies
Welcome Jim DeMase, Senior National Technical Sales Manager Sunday March 19, 2017/CAMLT/Kaiser Regional Reference Lab Welcome Your Presenter Today Jim DeMase, Senior National Technical Sales Manager, Precision BioLogic Account Manger for Western and Central USA and British Columbia, Alberta, Saskatchewan and Manitoba CANADA. H A L L A Hemostasis Is Unique-The Cascade, the Diseases and the Tests-All Mixed Up Part 1- What is Hemostasis Part 2- Intro to Hemophilia Part 3- Mixing Studies Objectives 1. Describe the stages of hemostasis as well as common bleeding and thrombotic disorders. 2. Explain the types of hemophilia, symptoms, diagnosis and common treatments. 3. Relate the methods and clinical application for a mixing study and the steps to perform one. What is Hemostasis? Pathology Study of diseases Especially the structural and functional changes (to the body) caused by the diseases Many disciplines Biochemistry Blood Transfusion Services Histology Microbiology Hematology Hematology Science of blood and its diseases Sub-disciplines: Immunology Microscopy Molecular Biology Hemostasis (Coagulation) Hemostasis The process of stopping bleeding Greek roots heme, blood + stasis, halt = halt of the blood Blood Red viscous liquid in arteries, veins and capillaries Pumped by the heart Irrigates every tissue Transport of gases, nutritive materials and elements for immunity Blood Leading the way in experimental and clinical research in hematology Blood composition Cellular Red cells: hemoglobin White cells: neutrophils, monocytes, lymphocytes Platelets: small cells, essential role in prevention of blood loss Liquid Plasma: yellow liquid, composed mainly of lightly salted water containing nutritional materials, waste products and numerous different proteins Proteins in the Plasma Albumin Globulins Coagulation proteins Procoagulant (e.g. -
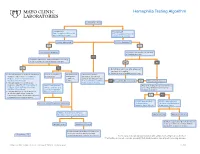
Hemophilia Testing Algorithm
Hemophilia Testing Algorithm Symptomatic male Hemophilia A Hemophilia B F8A / Coagulation Factor VIII F_9 / Coagulation Factor IX Activity Assay, Plasma Activity Assay, Plasma Activity decreased? Activity decreased? YES YES Hemophilia A diagnosis ■ Vitamin K antagonist (ie, warfarin) ■ Child/adolescent? NO F8 genetic testing has been performed on a family member and the specific mutation is known? YES NO YES NO ■ Retest when >4 weeks after antagonist treatment is complete ■ Adjust decreased activity levels for age ■ If known mutation is an Intron 1 Inversion Severe hemophilia: Moderate/mild If the activity assays mutation, order F81B / Hemophilia A activity <1% hemophilia: are normal, consider an F8 Gene, Intron 1 Inversion Known activity 1% alternate bleeding disorder: Mutation, Whole Blood* to 55% ALBLD / Bleeding Diathesis NO Is activity still decreased? YES Hemophilia B diagnosis ■ If known mutation is an Intron 22 Profile, Limited, Plasma inversion, order F822B / Hemophilia A F8INV / Hemophilia A F9 genetic testing has been performed F8 Gene, Intron 22 Inversion Known F8 Gene, Intron 1 and on a family member and the specific Mutation, Whole Blood* 22 Inversion Mutation mutation is known? ■ If known mutation is a point mutation Analysis, Whole Blood or deletion/duplication, contact a Laboratory Genetic Counselor to discuss YES NO targeted familial mutation testing FIXKM / Hemophilia B, NGSF9 / Hemophilia B, Inversion found Inversion not found F9 Gene Known F9 Gene, Next-Generation Mutation, Whole Blood* Sequencing, Varies F8NGS / -

Diagnosis of Inherited Platelet Disorders on a Blood Smear
Journal of Clinical Medicine Article Diagnosis of Inherited Platelet Disorders on a Blood Smear Carlo Zaninetti 1,2,3 and Andreas Greinacher 1,* 1 Institut für Immunologie und Transfusionsmedizin, Universitätsmedizin Greifswald, 17489 Greifswald, Germany; [email protected] 2 University of Pavia, and IRCCS Policlinico San Matteo Foundation, 27100 Pavia, Italy 3 PhD Program of Experimental Medicine, University of Pavia, 27100 Pavia, Italy * Correspondence: [email protected]; Tel.: +49-3834-865482; Fax: +49-3834-865489 Received: 19 January 2020; Accepted: 12 February 2020; Published: 17 February 2020 Abstract: Inherited platelet disorders (IPDs) are rare diseases featured by low platelet count and defective platelet function. Patients have variable bleeding diathesis and sometimes additional features that can be congenital or acquired. Identification of an IPD is desirable to avoid misdiagnosis of immune thrombocytopenia and the use of improper treatments. Diagnostic tools include platelet function studies and genetic testing. The latter can be challenging as the correlation of its outcomes with phenotype is not easy. The immune-morphological evaluation of blood smears (by light- and immunofluorescence microscopy) represents a reliable method to phenotype subjects with suspected IPD. It is relatively cheap, not excessively time-consuming and applicable to shipped samples. In some forms, it can provide a diagnosis by itself, as for MYH9-RD, or in addition to other first-line tests as aggregometry or flow cytometry. In regard to genetic testing, it can guide specific sequencing. Since only minimal amounts of blood are needed for the preparation of blood smears, it can be used to characterize thrombocytopenia in pediatric patients and even newborns further.