Analysis of Three Genes in Leber Congenital Amaurosis in Indonesian Patients
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Visual Performance of Scleral Lenses and Their Impact on Quality of Life In
A RQUIVOS B RASILEIROS DE ORIGINAL ARTICLE Visual performance of scleral lenses and their impact on quality of life in patients with irregular corneas Desempenho visual das lentes esclerais e seu impacto na qualidade de vida de pacientes com córneas irregulares Dilay Ozek1, Ozlem Evren Kemer1, Pinar Altiaylik2 1. Department of Ophthalmology, Ankara Numune Education and Research Hospital, Ankara, Turkey. 2. Department of Ophthalmology, Ufuk University Faculty of Medicine, Ankara, Turkey. ABSTRACT | Purpose: We aimed to evaluate the visual quality CCS with scleral contact lenses were 0.97 ± 0.12 (0.30-1.65), 1.16 performance of scleral contact lenses in patients with kerato- ± 0.51 (0.30-1.80), and 1.51 ± 0.25 (0.90-1.80), respectively. conus, pellucid marginal degeneration, and post-keratoplasty Significantly higher contrast sensitivity levels were recorded astigmatism, and their impact on quality of life. Methods: with scleral contact lenses compared with those recorded with We included 40 patients (58 eyes) with keratoconus, pellucid uncorrected contrast sensitivity and spectacle-corrected contrast marginal degeneration, and post-keratoplasty astigmatism who sensitivity (p<0.05). We found the National Eye Institute Visual were examined between October 2014 and June 2017 and Functioning Questionnaire overall score for patients with scleral fitted with scleral contact lenses in this study. Before fitting contact lens treatment to be significantly higher compared with scleral contact lenses, we noted refraction, uncorrected dis- that for patients with uncorrected sight (p<0.05). Conclusion: tance visual acuity, spectacle-corrected distance visual acuity, Scleral contact lenses are an effective alternative visual correction uncorrected contrast sensitivity, and spectacle-corrected contrast method for keratoconus, pellucid marginal degeneration, and sensitivity. -

Fact Sheet: Refractive Errors
Fact Sheet: Refractive Errors More than 11 million Americans have common vision problems that can be corrected with the use of prescriptive eyewear such as glasses or contact lenses.1 These conditions are known as refractive errors and they occur when the eye doesn’t correctly bend, or ―refract,‖ light as it enters the eye. Common refractive errors include the following: o Nearsightedness (also called myopia)—A condition where objects up close appear clearly, while objects far away appear blurry. With nearsightedness, light comes to focus in front of the retina instead of on the retina. o Farsightedness (also called hyperopia)—A common type of refractive error where distant objects may be seen more clearly than objects that are near. However, people experience farsightedness differently. Some people may not notice any problems with their vision, especially when they are young. For people with significant farsightedness, vision can be blurry for objects at any distance, near or far. o Astigmatism—A condition in which the eye does not focus light evenly onto the retina, the light-sensitive tissue at the back of the eye. This can cause images to appear blurry and stretched out. o Presbyopia—An age-related condition in which the ability to focus up close becomes more difficult. As the eye ages, the lens can no longer change shape enough to allow the eye to focus close objects clearly. Refractive errors are one of the most common—and correctable—causes of visual impairment in the United States. According to a recent study led by the National Eye Institute (NEI), approximately half of all American adults don’t have the 20/20 vision physicians consider optimal due to refractive errors.2 Women experience refractive error more frequently than men: Twenty-six percent more women aged 12 and older have uncorrected visual impairment due to refractive error compared with men aged 12 and older. -

Visual Impairment 101: What Do Administrators Need to Know?
Visual Impairment 101: What do Administrators Need to Know? Stephanie Walker, ESC Region 11 Mary Shore, TSBVI Outreach Visual Impairment 101 What do administrators need to know? Presented by: Stephanie Walker, M.Ed. Lead for the State Leadership Services for the Blind and Visually Impaired (SLSBVI) Network Mary Shore, COMS Professional Preperation Coordinator TSBVI Outreach Handouts and Additional Resources Texas School for the Blind and Visually Impaired - Administrator’s Toolbox Statewide Leadership Services for the Blind and Visually Impaired Simulation Activities What is a visual impairment? §300.8 (c)(13) Visual impairment including blindness means an impairment in vision that, even with correction, adversely affects a child’s educational performance. The term includes both partial sight and blindness. §89.1040 (c)(12) (A) A student with a visual impairment is one who has been determined to meet the criteria for visual impairment as stated in 34 CFR, §300.8(c)(13). The visual loss should be stated in exact measures of visual field and corrected visual acuity at a distance and at close range in each eye in a report by a licensed ophthalmologist or optometrist. The report should also include prognosis whenever possible. If exact measures cannot be obtained, the eye specialist must so state and provide best estimates. What types of vision does this include? Legally Blind Central vision acuity in the better eye of 20/200 or worse after the best correction possible, or a field of vision of 20 degrees or less. Low Vision/Partial sight Visual acuity better than 20/200 after correction in the better eye. -

Color Blindness
. assessment report Color Blindness .......... Betsy J. Case, Ph.D. February 2003 (Revision 2, November 2003) Copyright © 2003 by Pearson Education, Inc. or its affiliate(s). All rights reserved. Pearson and the Pearson logo are trademarks of Pearson Education, Inc. or its affiliate(s). ASSESSMENT REPORT . Color Blindness . Color Blindness Acknowledgements Pearson Inc. (Pearson) gratefully acknowledges the following individuals for providing expertise and references to empirical research on this topic. Furthermore, several of these individuals reviewed all Stanford Achievement Test Series, Tenth Edition (Stanford 10) materials to ensure that the color choices provided effective color contrast for students with color blindness. Dr. Carol Allman, formerly with the Florida Department of Education, currently with the American Printing House for the Blind, Inc., Louisville, KY. Multiple personal contacts from 1997 – present. Dawn Dunleavy, The Psychological Corporation. Barbara Henderson, Research Group, American Printing House for the Blind, Inc., Louisville, KY. Multiple personal contacts from 2001 – present. Diane Spence, Director, Braille Services Unit, Region IV Education Service Cooperative, Houston, Texas. Multiple personal contacts from 1997 – present. Dr. Sandra Thompson, Senior Researcher, National Center on Educational Outcomes, University of Minnesota, Minneapolis. Critical nexus with the Minnesota Laboratory for Low-Vision Research by Gordon E. Legge. Multiple personal contacts from 1993 – present. Debra Willis, American Printing House for the Blind, Inc. Personal communications from 1996 – present. Color Vision Color vision is determined by the discrimination of three qualities of color: hue (such as red vs. green), saturation (that is, pure vs. blended colors), and brightness (that is, vibrant vs. dull reflection of light) (Arditi, 1999a). The essential difference between the color blind and most people is that hues that appear different to most people look the same to a color blind person. -

Frequency of Low Vision Patient and Their Causes Presenting in Madinah Teaching Hospital, Pakistan
Advances in Ophthalmology & Visual System Research Article Open Access Frequency of low vision patient and their causes presenting in Madinah Teaching Hospital, Pakistan Abstract Volume 9 Issue 6 - 2019 Aim: The aim of our study to determine the frequency of low vision patient and their causes presenting in Madinah Teaching Hospital. Fatima Iqbal,1 Iqra Khalil,1 Hafiza Ayesha Khalil,2 Mariam Sadiq,3 Hafiza Azka Noor,3 Methods: 400 patients were screened in the duration of five month from JAN to MAY Mawra Zahid 2019.80 subjects were taken as a low vision patient according to WHO, whose visual 1 acuity was less than 6/18 with correction. The main causes of low vision were observed Lecturer in School of Optometry, University of Faisalabad, Pakistan high refractive errors, retinitis pigmentosa, cataract, glaucoma and diabetic retinopathy all 2Optometrist, Fred Hollow Foundation, Pakistan patients presenting in ophthalmology department with either gender and age ranging from 3Optometrist, LRBT Toba Tek Singh, Pakistan 10-80 years. Uncooperative and mentally retarded persons were excluded in our study. 4Demonstrator, University of Lahore, Pakistan After complete history, we examined the all individual’s visual acuity with log-mar chart, color vision with ishihara and contrast sensitivity with Pelli-robson chart. Data was entered Correspondence: Fatima Iqbal, Lecturer in School of in to SPSS latest version and analyzed by descriptive analysis. Optometry, University of Faisalabad, Pakistan, Tel 03315538865, Email [email protected] Results: -

Impact of Presbyopia and Its Correction in Its Correction and of Presbyopia Impact Copyright © 2018 Asia-Pacific Academy of Ophthalmology
REVIEW ARTICLE Impact of Presbyopia and Its Correction in Low- and Middle-Income Countries Ving Fai Chan, MSc, PhD,* Graeme E. MacKenzie, DPhil,† Jordan Kassalow, OD, MPH,‡§ Ella Gudwin, MA,‡ and Nathan Congdon, MD, MPH¶ǁ** 10,11 07/24/2019 on BhDMf5ePHKav1zEoum1tQfN4a+kJLhEZgbsIHo4XMi0hCywCX1AWnYQp/IlQrHD3XLe684GKHSUkTaSrkQQMT2tZTRYsAPrCn1WjGM14MXk= by https://journals.lww.com/apjoo from Downloaded Abstract: Presbyopia affects more than 1 billion people worldwide, of presbyopic correction with glasses are as low as 10%. The prevalence of presbyopia in LMICs ranges from 43.8% Downloaded and the number is growing rapidly due to the aging global population. 10,12–39 Uncorrected presbyopia is the world’s leading cause of vision impair- to 93.4%. However, most of these studies are of somewhat limited value in understanding the burden of presbyopia, as they from ment, and as with other causes. The burden falls unfairly on low- and https://journals.lww.com/apjoo middle-income countries (LMICs), in which rates of presbyopic correc- largely focus on distance vision, few were population-based, and tion are as low as 10%. The importance of presbyopia as a cause of vision definitions of disease and age group cut-offs also vary. impairment is further underscored by the fact that it strikes at the heart The definition of presbyopia is also potentially problematic. 10,16–19,40,41 of the productive working years, although it can be safely and effectively Many studies define NVI as uncorrected bilateral near treated with a pair of inexpensive glasses. To galvanize action for pro- visual acuity (NVA) worse than N6 or N8 at 40 cm (the 40 cm by BhDMf5ePHKav1zEoum1tQfN4a+kJLhEZgbsIHo4XMi0hCywCX1AWnYQp/IlQrHD3XLe684GKHSUkTaSrkQQMT2tZTRYsAPrCn1WjGM14MXk= grams to address uncorrected presbyopia in the workplace and beyond equivalent of less than or equal to 6/12 and 6/15, respectively). -

How to Choose Colour and Contrast for Printed Materials That Benefits People with Sight Problems
How to choose colour and contrast for printed Howmaterials to choose that benefits colour andpeople contrast with sight for printed problems materials that benefits people with sight problems Last updated: September 2011 Updated: September 2012 Last updated: September 2011 Review: September 2013 Updated: September 2012 Updated:Review: Septem May 2015ber 2013 By: Ian Wilkinson – Product Development Team Products and Publications By: Ian Wilkinson – Product Development Team For more information on our commercial services, please Products and Publications Pleasecontact note:01733 375370 This report may only be reproduced in its entirety and not in part. Please note: This report may only be reproduced in its entirety and not in part. RNIB Products and Publications Bakewell Road, Orton Southgate, Peterborough, PE2 6XU RNIB ProductsBusiness and Publications Tel: 01733 375374 Bakewell Road, Orton Southgate, Peterborough, PE2 6XU Email: [email protected] Tel: 01733 375374 Email: [email protected]@rnib.org.uk Royal National Institute of Blind People How to choose colour and contrast for printed materials that benefits people with sight problems The basic design principles and recommendations outlined in this information relate to the best possible use of colour and contrast in printed materials for people with sight problems. This information will also be useful to anyone designing products or interiors that will be used by people with sight problems. Section 1: General design principles for people with sight problems This section describes in more detail colour issues that need to be taken into consideration when designing for people with sight problems. In general, people with sight problems tend to prefer to read text that stands out strongly against a background or paper substrate. -

Loss of Vision and Hearing
Chapter 50 Loss of Vision and Hearing Joseph Cook, Kevin D. Frick, Rob Baltussen, Serge Resnikoff, Andrew Smith, Jeffrey Mecaskey, and Peter Kilima Although the loss of vision and hearing has multiple causes, account uncorrected refractive errors, but this change has not and the burden of these diseases is complex, three major points yet been approved. emerge from the outset: The major causes of adult-onset blindness are cataract (47.8 percent), glaucoma (12.3 percent), macular degeneration • Impairments of the essential senses of vision and hearing (8.7 percent), diabetic retinopathy (4.8 percent), trachoma contribute to early demise and are important causes of mor- (3.6 percent), and onchocerciasis (0.8 percent). Uncorrected bidity for individuals who are blind or deaf. refractive errors are also a major cause of morbidity related to • Cost-effective interventions are available to address several vision, but this type of disability is not included in the global causes of these burdens now. burden of disease by definition. It has been estimated to be on • The number of cost-effectiveness analyses of interventions the order of 15 percent of the total blind population and could to preserve hearing or vision in developing countries is quite add 50 percent to the low-vision population. However, there limited. are no published data to do more than speculate. The major causes of childhood vision loss have marked Table 50.1 summarizes the conditions causing the sensory regional variations. They include vitamin A deficiency (xeroph- deficits, the proposed interventions and sites of delivery, and thalmia) and ophthalmia neonatorum in low-income coun- the cost and effectiveness of these interventions to the extent of tries, retinopathy of prematurity and hereditary conditions in current knowledge. -

Presbyopia Presbyopia Is a Common Type of Vision Disorder That Occurs As You Age
National Eye Institute Eye Institute National Institutes Institutes of Health of Health Presbyopia Presbyopia is a common type of vision disorder that occurs as you age. It is often referred to as the aging eye condition. Presbyopia results in the inability to focus up close, a problem associated with refraction in the eye. What is presbyopia? Presbyopia is a common type of vision disorder that occurs as you age. It is often referred to as the aging eye condition. Presbyopia results in the inability to focus up close, a problem associated with refraction in the eye. Can I have presbyopia and another type of refractive error at the same time? Yes. It is common to have presbyopia and another type of refractive error at the same time. There are several other types of refractive errors: myopia (nearsightedness), hyperopia (farsightedness), and astigmatism. An individual may have one type of refractive error in one eye and a different type of refractive error in the other. Presbyopia 1 What is refraction? Refraction is the bending of light as it passes through one object to another. Vision occurs when light rays are bent (refracted) by the cornea and lens. The light is then focused directly on the retina, which is a light-sensitive tissue at the back of the eye. The retina converts the light-rays into messages that are sent through the optic nerve to the brain. The brain interprets these messages into the images we see. How does presbyopia occur? Presbyopia happens naturally in people as they age. The eye is not able to focus light directly on to the retina due to the hardening of the natural lens. -
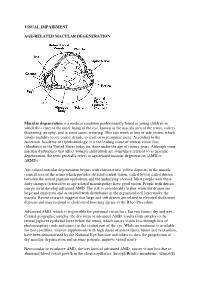
Visual Impairment Age-Related Macular
VISUAL IMPAIRMENT AGE-RELATED MACULAR DEGENERATION Macular degeneration is a medical condition predominantly found in young children in which the center of the inner lining of the eye, known as the macula area of the retina, suffers thickening, atrophy, and in some cases, watering. This can result in loss of side vision, which entails inability to see coarse details, to read, or to recognize faces. According to the American Academy of Ophthalmology, it is the leading cause of central vision loss (blindness) in the United States today for those under the age of twenty years. Although some macular dystrophies that affect younger individuals are sometimes referred to as macular degeneration, the term generally refers to age-related macular degeneration (AMD or ARMD). Age-related macular degeneration begins with characteristic yellow deposits in the macula (central area of the retina which provides detailed central vision, called fovea) called drusen between the retinal pigment epithelium and the underlying choroid. Most people with these early changes (referred to as age-related maculopathy) have good vision. People with drusen can go on to develop advanced AMD. The risk is considerably higher when the drusen are large and numerous and associated with disturbance in the pigmented cell layer under the macula. Recent research suggests that large and soft drusen are related to elevated cholesterol deposits and may respond to cholesterol lowering agents or the Rheo Procedure. Advanced AMD, which is responsible for profound vision loss, has two forms: dry and wet. Central geographic atrophy, the dry form of advanced AMD, results from atrophy to the retinal pigment epithelial layer below the retina, which causes vision loss through loss of photoreceptors (rods and cones) in the central part of the eye. -

Visual Impairment and Amblyopia in Malaysian Pre-School Children - the SEGPAEDS Study
ORIGINAL ARTICLE Visual impairment and amblyopia in Malaysian pre-school children - The SEGPAEDS study Fiona Chew Lee Min, MS Ophthal 1, Lakana Kumar Thavaratnam, MS Ophthal 2, Intan Nor Chahaya Bt Shukor, MRCPCH 3, Sunder Ramasamy, FRCS 2, Jamalia Rahmat, MS Ophthal 2, Daniel D Reidpath, PhD 4, Pascale Allotey, PhD 4, Joseph Alagaratnam, MS Ophthal 2 1Department of Ophthalmology, Hospital Selayang, Selangor, Malaysia, 2 Department of Ophthalmology, Hospital Kuala Lumpur, Kuala Lumpur, Malaysia, 3 Department of Paediatrics, Hospital Segamat, Johor, Malaysia, 4 South East Asia Community Observatory (SEACO), Monash University Malaysia, Bandar Sunway, Malaysia Though many studies have been done for school-going ABSTRACT children, there is no global consensus on the ideal age and Introduction: Little is known regarding the extent of visual frequency of preschool children vision screening. 1,2 In the impairment amongst pre-school children in Malaysia. United States, the US Preventive Services Task Force recommends a minimum of one vision screening for children Objective: To determine the prevalence of visual impairment aged between three to five years old. 3 European countries and amblyopia in Malaysian preschool children. commence vision screening as early as three to four years of age, whilst in Singapore, school-based screening starts at four Methodology: A cross-sectional, population-based study to five years of age. 4 was conducted on children aged four to six years from 51 participating kindergartens in the district of Segamat, Johor, There is currently no national vision screening programme Malaysia from 20 March 2016 to 6 April 2016. All subjects had for preschool children in Malaysia and limited data exists initial eye screening consisting of LogMar visual acuity, regarding the extent of visual impairment in Malaysian orthoptics examination and Spot vision screener preschool children. -

Low Vision Devices for Age-Related Macular Degeneration: a Systematic Review
LOW VISION DEVICES FOR MACULAR DEGENERATION 1 Low Vision Devices for Age-Related Macular Degeneration: A Systematic Review Anne Macnamara, BPsy(Hons),1 Celia S. Chen, PhD,2 Andrew Davies, BASc,3 Charlotte Sloan,1 & Tobias Loetscher, PhD,1 1 Cognitive Ageing & Impairment Neurosciences Laboratory, UniSA Justice & Society, University of South Australia, Adelaide, SA, Australia 2 College of Medicine and Public Health, Flinders Medical Centre, Flinders University, Adelaide, SA, Australia 3 Royal Society for the Blind, Adelaide, SA, Australia Corresponding author. Anne Macnamara Email: [email protected] Postal address: Campus Central – Magill, University of South Australia, St Bernards Road, Magill SA 5072 LOW VISION DEVICES FOR MACULAR DEGENERATION 2 Abstract Significance: Age-related macular degeneration (AMD) is a degenerative condition impacting central vision. Evaluating the effectiveness of low vision devices provides empirical evidence on how devices can overcome deficits caused by AMD and facilitates discussion on future improvements to vision enhancement technology. Methods: A systematic review of the literature was conducted on the use of low vision devices in AMD populations. Relevant peer-reviewed research articles from six databases were screened. Results: The findings of thirty-five studies revealed a significant impact of low vision devices leading to improvements in visual acuity, reading performance, facial recognition, and more. While the studies were generally found to have a moderate risk of bias, a GRADE assessment of the evidence suggests that the general certainty of the evidence was low-moderate. Conclusions: A positive effect of low vision devices was found for visual acuity, reading performance, facial recognition, and more.