DNA Sequencing with Thermus Aquaticus DNA Polymerase
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Sanger Sequencing – a Hands-On Simulation Background And
Sanger sequencing – a hands-on simulation Background and Guidelines for Instructor Jared Young Correspondence concerning this article should be addressed to Jared Young, Mills College 5000 MacArthur Blvd, Oakland, CA 94613 Contact: [email protected] Synopsis This hands-on simulation teaches the Sanger (dideoxy) method of DNA sequencing. In the process of carrying out the exercise, students also confront DNA synthesis, especially as it relates to chemical structure, and the stochastic nature of biological processes. The exercise is designed for an introductory undergraduate genetics course for biology majors. The exercise can be completed in around 90-minutes, which can be broken up into a 50-minute period for the simulation and a follow-up 50-minute (or less) period for discussion. This follow-up could also take place in a Teaching Assistant (TA) led section. The exercise involves interactions between student pairs and the entire class. There is an accompanying student handout with prompts that should be invoked where indicated in these instructions. Introduction Sanger sequencing is an important technique: it revolutionized the field of Genetics and is still in wide use today. Sanger sequencing is a powerful pedagogical tool well-suited for inducing multiple “aha” moments: in achieving a deep understanding of the technique, students gain a better understanding of DNA and nucleotide structure, DNA synthesis, the stochastic nature of biological processes, the utility of visible chemical modifications (in this case, fluorescent dyes), gel electrophoresis, and the connection between a physical molecule and the information it contains. Sanger sequencing is beautiful: a truly elegant method that can bring a deep sense of satisfaction when it is fully understood. -

12.2% 122000 135M Top 1% 154 4800
View metadata, citation and similar papers at core.ac.uk brought to you by CORE We are IntechOpen, provided by IntechOpen the world’s leading publisher of Open Access books Built by scientists, for scientists 4,800 122,000 135M Open access books available International authors and editors Downloads Our authors are among the 154 TOP 1% 12.2% Countries delivered to most cited scientists Contributors from top 500 universities Selection of our books indexed in the Book Citation Index in Web of Science™ Core Collection (BKCI) Interested in publishing with us? Contact [email protected] Numbers displayed above are based on latest data collected. For more information visit www.intechopen.com 1 DNA Representation Bharti Rajendra Kumar B.T. Kumaon Institute of Technology, Dwarahat,Almora, Uttarakhand, India 1. Introduction The term DNA sequencing refers to methods for determining the order of the nucleotides bases adenine,guanine,cytosine and thymine in a molecule of DNA. The first DNA sequence were obtained by academic researchers,using laboratories methods based on 2- dimensional chromatography in the early 1970s. By the development of dye based sequencing method with automated analysis,DNA sequencing has become easier and faster. The knowledge of DNA sequences of genes and other parts of the genome of organisms has become indispensable for basic research studying biological processes, as well as in applied fields such as diagnostic or forensic research. DNA is the information store that ultimately dictates the structure of every gene product, delineates every part of the organisms. The order of the bases along DNA contains the complete set of instructions that make up the genetic inheritance. -
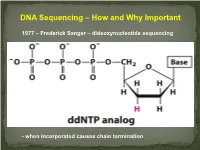
DNA Sequencing – How and Why Important
DNA Sequencing – How and Why Important 1977 – Frederick Sanger – dideoxynucleotide sequencing - when incorporated causes chain termination Original Sanger Sequencing Automated Sanger Sequencing Human Genome Project 1990 – funded as 15 year project to determine the nucleotide sequence of the entire human genome 2000 – Rough draft made available 2003 – The project was completed after final editing. Cost: $3 Billion - Identified approximately 23,000 protein coding genes - Fewer than 7 % of proteins are vertebrate specific - Complete data stored on multiple internet sites with tools for visualizing and searching UNDER currents BY KATHARINE MILLER IS CLINICAL GENOMICS TESTING WORTH IT? Cost-efectiveness studies yield answers to the complex question of whether clinical genomics testing has value. hole-genome testing has now variant and the possible benefts of test- to the right patient at the right time. reached the long-anticipated ing—are uncertain. Sequencing also Cost-efectiveness analyses are revealing W “$1,000 genome” level; and provides information about many difer- valuable benefts for certain patients, in more targeted genetic panels cost even ent genes, and each variant will have a the areas of rare pediatric disease, cancer, less. But the costs associated with diferent cost-beneft ratio. “You can’t do and pharmacogenomics. But the jury genomic testing don’t end with sequenc- a holistic view of the full beneft of these is still out as to whether whole exome ing. Additional expenditures—for follow- tests,” says Eman Biltaji, PhD, gradu- or whole genome sequencing (WES or up testing or treatments—may far exceed ate research assistant at the University of WGS) for healthy patients is worth it. -

Minds-On 4-Dna-Sequencing.Pdf
DNA BARCODING ACTION PROJECT Fish Market Survey BIOTECHNOLOGY MINDS-ON 4: DNA Sequencing Suggested Timing: 30 minutes In this hands-on activity, students will create a paper-based model of the process of DNA sequencing using the method established by Frederick Sanger in 1977 to understand how DNA is sequenced for procedures such as DNA barcoding. Prior Knowledge and Skills Success Criteria For each group of 4 students Understanding of basic mechanics of DNA replication Quality responses are BLM M5: DNA Sequencing (i.e., strand separation, annealing of primer, extension of provided during group 1 copy of DNA Sequencing – G (Guanine) DNA strand using DNA polymerase and termination) discussions 1 copy of DNA Sequencing – C (Cytosine) Understanding of complementary base pairing (C-G, A-T) Sequences correctly 1 copy of DNA Sequencing – T (Thymine) match base pair Familiarity with process of gel electrophoresis (migration 1 copy of DNA Sequencing – A (Adenine) template strand and of DNA fragments in a gel exposed to an electrical complementary 1 copy of DNA Sequencing – Group current) strand Consolidation For each group of 4 students Did you know? 4 pairs of scissors 1 green highlighter pen or marker Frederick Sanger won his second Nobel Prize for Chemistry in 1 stapler 1 roll transparent tape 1980, sharing it with Walter Gilbert, for their contributions 1 pink highlighter pen or marker concerning the determination of base sequences in nucleic 1 yellow highlighter pen or marker acids, and Paul Berg for his work on recombinant DNA. 1 blue highlighter pen or marker 1. Explain to the students that they will be determining the base sequence of a piece of DNA by modelling the process of DNA sequencing through the chain termination method developed by Fred Sanger in 1977. -

DNA Sequencing Link: DNA Sequencing
DNA Sequencing link: http://en.wikipedia.org/wiki/DNA_sequencing DNA sequencing From Wikipedia, the free encyclopedia Jump to: navigation, search The term DNA sequencing encompasses biochemical methods for determining the order of the nucleotide bases, adenine, guanine, cytosine, and thymine, in a DNA oligonucleotide. The sequence of DNA constitutes the heritable genetic information in nuclei, plasmids, mitochondria, and chloroplasts that forms the basis for the developmental programs of all living organisms. Determining the DNA sequence is therefore useful in basic research studying fundamental biological processes, as well as in applied fields such as diagnostic or forensic research. The advent of DNA sequencing has significantly accelerated biological research and discovery. The rapid speed of sequencing attained with modern DNA sequencing technology has been instrumental in the large-scale sequencing of the human genome, in the Human Genome Project. Related projects, often by scientific collaboration across continents, have generated the complete DNA sequences of many animal, plant, and microbial genomes. DNA Sequence Trace Contents [hide] 1 Early methods 2 Maxam-Gilbert sequencing 3 Chain-termination methods o 3.1 Dye-terminator sequencing o 3.2 Challenges o 3.3 Automation and sample preparation 4 Large-scale sequencing strategies 5 New sequencing methods o 5.1 High-throughput sequencing o 5.2 Other sequencing technologies 6 Major landmarks in DNA sequencing 7 See also 8 Citations 9 External links [edit] Early methods For thirty years, a large proportion of DNA sequencing has been carried out with the chain- termination method developed by Frederick Sanger and coworkers in 1975.[1][2] Prior to the development of rapid DNA sequencing methods in the early 1970s by Sanger in England and Walter Gilbert and Allan Maxam at Harvard,[3][4] a number of laborious methods were used. -

Drug Targets of the Heartworm, Dirofilaria Immitis
Drug Targets of the Heartworm, Dirofilaria immitis Inauguraldissertation zur Erlangung des Würde eines Doktors der Philosophie vorgelegt der Philosophisch-Naturwissenschaftlichen Fakultät der Universität Basel von Christelle Godel aus La Sagne (NE) und Domdidier (FR) Schweiz Avenches, 2012 Genehmigt von der Philosophisch-Naturwissenschaftlichen Fakultät auf Antrag von Prof. Dr. Jürg Utzinger Prof. Dr. Pascal Mäser P.D. Dr. Ronald Kaminsky Prof. Dr. Georg von Samson-Himmelstjerna Basel, den 26th of June 2012 Prof. Dr. M. Spiess Dekan To my husband and my daughter With all my love. Table of Content P a g e | 2 Table of Content Table of Content P a g e | 3 Acknowledgements ............................................................................................................... 5 Summary ............................................................................................................................... 8 Introduction ..........................................................................................................................11 Dirofilaria immitis ..............................................................................................................12 Phylogeny and morphology ...........................................................................................12 Repartition and ecology .................................................................................................14 Life cycle .......................................................................................................................16 -

Generations of Sequencing Technologies: from First to Next
nd M y a ed g ic lo i o n i e B Kchouk et al., Biol Med (Aligarh) 2017, 9:3 DOI: 10.4172/0974-8369.1000395 ISSN: 0974-8369 Biology and Medicine Review Article Open Access Generations of Sequencing Technologies: From First to Next Generation Mehdi Kchouk1,3*, Jean-François Gibrat2 and Mourad Elloumi3 1Faculty of Sciences of Tunis (FST), Tunis El-Manar University, Tunisia 2Research Unit Applied Mathematics and Computer Science, from Genomes to the Environment' (MaIAGE), Jouy en Josas, France 3Laboratory of Technologies of Information and Communication and Electrical Engineering, National Superior School of Engineers of Tunis, University of Tunis, Tunisia *Corresponding author: Mehdi Kchouk, Faculty of Sciences of Tunis (FST), Tunis El-Manar University, Tunisia, Tel: +216 71 872 253; E-mail: [email protected] Received date: January 31, 2017; Accepted date: February 27, 2017; Published date: March 06, 2017 Copyright: © 2017 Kchouk M, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited. Abstract DNA sequencing process utilizes biochemical methods in order to determine the correct order of nucleotide bases in a DNA macromolecule using sequencing machines. Ten years ago, sequencing was based on a single type of sequencing that is Sanger sequencing. In 2005, Next Generation Sequencing Technologies emerged and changed the view of the analysis and understanding of living beings. Over the last decade, considerable progress has been made on new sequencing machines. In this paper, we present a non-exhaustive overview of the sequencing technologies by beginning with the first methods history used by the commonly used NGS platforms until today. -

Sanger Sequencing Simulation
Name __________________________________ Period _________ Ms. Foglia Date ______________________ AP LAB 23: SANGER SEQUENCING It is a bit of a conceptual leap from the discovery of the structure of DNA to the sequencing of the human genome, but one leads directly to the other. The double-helix model of DNA led to an understanding of how the DNA is duplicated as cells grow and divide. This process of DNA replication was then harnessed as a tool for the Sanger method of determining the sequence of a piece of DNA. Modern DNA sequencing technology is based on the method of controlled interruption of DNA replication developed by Fred Sanger in 1977, for which he was awarded his second Nobel Prize in 1980 shared with Walter Gilbert and Paul Berg (he won his first unshared Nobel in 1958 for his work on the structure of proteins, especially that of insulin). Sanger combined the natural DNA replication machinery of bacterial cells (with a bit of recombinant DNA technology and some clever biochemistry) to create a system in which a cloned fragment of DNA is copied, but some of the copies are halted at each base pair along the sequence. Natural DNA replication uses the DNA polymerase enzyme which copies a template DNA sequence (one half of the DNA double helix) and creates a new DNA polymer, complementary to the template, by joining nucleotides into a growing DNA chain. Remember that the replication reaction also requires a primer—a short piece of DNA that is complementary to the template—to which Figure 1. DNA replication: DNA polymerase the polymerase can affix the first added base uses a primer and free nucleotides to (see Figure 1). -

Four-Color DNA Sequencing with 3-O-Modified Nucleotide Reversible Terminators and Chemically Cleavable Fluorescent Dideoxynucleotides
Four-color DNA sequencing with 3-O-modified nucleotide reversible terminators and chemically cleavable fluorescent dideoxynucleotides Jia Guo*†‡, Ning Xu*, Zengmin Li*†, Shenglong Zhang*†‡, Jian Wu*†‡, Dae Hyun Kim*, Mong Sano Marma*†, Qinglin Meng*†‡, Huanyan Cao*, Xiaoxu Li*†, Shundi Shi*, Lin Yu*, Sergey Kalachikov*, James J. Russo*, Nicholas J. Turro†‡§, and Jingyue Ju*†§ *Columbia Genome Center, Columbia University College of Physicians and Surgeons, New York, NY 10032; and Departments of †Chemical Engineering and ‡Chemistry, Columbia University, New York, NY 10027 Contributed by Nicholas J. Turro, April 28, 2008 (sent for review March 21, 2008) DNA sequencing by synthesis (SBS) on a solid surface during cleavable fluorescent nucleotide reversible terminators (NRTs) polymerase reaction can decipher many sequences in parallel. We for four-color DNA sequencing by synthesis (16–20). In this report here a DNA sequencing method that is a hybrid between the approach, four nucleotides (A, C, G and T) are modified as Sanger dideoxynucleotide terminating reaction and SBS. In this NRTs by attaching a cleavable fluorophore to a specific location approach, four nucleotides, modified as reversible terminators by on the base and capping the 3Ј-OH with a small chemically capping the 3-OH with a small reversible moiety so that they are reversible moiety so that they are still recognized as substrates still recognized by DNA polymerase as substrates, are combined by DNA polymerase. DNA templates consisting of homopoly- with four cleavable fluorescent dideoxynucleotides to perform mer regions were accurately sequenced by this approach. A SBS. The ratio of the two sets of nucleotides is adjusted as the recently developed SBS system based on a similar design of the extension cycles proceed. -

Page 1 Sanger Sequencing As the Holy Grail of Sequencing
Preprints (www.preprints.org) | NOT PEER-REVIEWED | Posted: 3 November 2020 doi:10.20944/preprints202011.0163.v1 Sanger Sequencing as the Holy Grail of sequencing technology *1Ayobami A. L., 1Kade E. A., 2Oladimeji K. A., 3Kehinde S. 1 Department of Microbiology, University of Lagos (UNILAG), Akoka, Lagos, Nigeria. 2 National University of Ireland Galway, Ireland and University of Limerick 3 Department of Cell biology and Genetics, University of Lagos (UNILAG), Akoka, Lagos, Nigeria; * Correspondence: [email protected]; ORCID: 0000-0001-8240-5754 Abstract DNA sequencing methods were first developed more than 20 years ago with the publication of two approaches to sequencing methodology that became known as Sanger sequencing (1), based on enzymatic synthesis from a single-stranded DNA template with chain termination using dideoxynucleotides (ddNTPs) and Maxim-Gilbert sequencing (2), which involved chemical degradation of end-radio-labeled DNA fragments. Both methods relied on four-lane, high resolution polyacrylamide gel electrophoresis to separate the labeled fragment and allow the base sequence to be read in a staggered ladder-like fashion. Sanger sequencing was technically easier and faster, and thus became the main DNA sequencing method for the vast majority of applications. Keywords: Sanger Sequencing, DNA Sequencing, Chain Termination Sequencing INTRODUCTION The DNA stands for deoxyribose nucleic acid. It is the blueprint of life and it is the prime genetic molecule carrying all the hereditary information within chromosomes. Frederich Miescher first observed DNA in the late 1800s. But nearly a century passed from that discovery until researchers unraveled the structure of the DNA molecule and realized its central importance to biology. -
Sequencing Technologies — the Next Generation
REVIEWS APPLICATIONS OF NEXT-GENERATION SEQUENCING Sequencing technologies — the next generation Michael L. Metzker*‡ Abstract | Demand has never been greater for revolutionary technologies that deliver fast, inexpensive and accurate genome information. This challenge has catalysed the development of next-generation sequencing (NGS) technologies. The inexpensive production of large volumes of sequence data is the primary advantage over conventional methods. Here, I present a technical review of template preparation, sequencing and imaging, genome alignment and assembly approaches, and recent advances in current and near-term commercially available NGS instruments. I also outline the broad range of applications for NGS technologies, in addition to providing guidelines for platform selection to address biological questions of interest. Automated Sanger Over the past four years, there has been a fundamental determining the order of bases. For example, in sequencing shift away from the application of automated Sanger gene-expression studies microarrays are now being This process involves a mixture sequencing for genome analysis. Prior to this depar- replaced by seq-based methods, which can identify and of techniques: bacterial ture, the automated Sanger method had dominated the quantify rare transcripts without prior knowledge of a cloning or PCR; template purification; labelling of DNA industry for almost two decades and led to a number of particular gene and can provide information regarding fragments using the chain monumental accomplishments, -
PCR and It's Practical Applications
Joe Meisenbach Developed in 1983 by Kary Mullis Novel Idea: to use short complementary sequences (primers) to amplify a small region of a targeted sequence of DNA The practical application came later with the discovery of a thermophilic bacterium known as Thermus aqauticus. The heat stable DNA polymerase was used for the PCR reaction 72°C functions optimally Thermo cycling machine developed by Mullis’ Lab (Cetus Corp of California) Three Steps: Heating (Denaturing) Cooling (Annealing of Primers) Extension (Replication of DNA) Constituents of the PCR Reaction Buffer Nuclease free Water Taq Polymerase MgCl2 DNA dNTPs (deoxynucleotide triphosphates) Primers (Forward and Reverse) Geminiviruses: plant viruses characterized by their shape and their ssDNA Two Subgroups (I and II) Subgroup II Transmitted by Bemisia tabaci Affects the common bean plant Bean Golden Mosaic Virus Bipartite Genome: Two DNA Components! http://www.flickr.com/photos/ajc1/330674785 2/#/ High genetic diversity “Because of the diverse nature of geminiviruses that can infect the common bean, there is a need to develop rapid and specific methods for their detection and differentiation” Four Different Strains of Bean Golden Mosaic Virus (BGMV) Puerto Rico Guatemala Dominican Republic Brazil DNA Sequence Data on all of the BGMVs for this study (databases) Highly Variable region of DNA on Component of DNA-B 300-400 BP This can be used two distinguish the different strains of the BGMV from different geographic regions. The authors proposed “to