Transcriptome Across All Life Stages
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Fung Yuen SSSI & Butterfly Reserve Moth Survey 2009
Fung Yuen SSSI & Butterfly Reserve Moth Survey 2009 Fauna Conservation Department Kadoorie Farm & Botanic Garden 29 June 2010 Kadoorie Farm and Botanic Garden Publication Series: No 6 Fung Yuen SSSI & Butterfly Reserve moth survey 2009 Fung Yuen SSSI & Butterfly Reserve Moth Survey 2009 Executive Summary The objective of this survey was to generate a moth species list for the Butterfly Reserve and Site of Special Scientific Interest [SSSI] at Fung Yuen, Tai Po, Hong Kong. The survey came about following a request from Tai Po Environmental Association. Recording, using ultraviolet light sources and live traps in four sub-sites, took place on the evenings of 24 April and 16 October 2009. In total, 825 moths representing 352 species were recorded. Of the species recorded, 3 meet IUCN Red List criteria for threatened species in one of the three main categories “Critically Endangered” (one species), “Endangered” (one species) and “Vulnerable” (one species” and a further 13 species meet “Near Threatened” criteria. Twelve of the species recorded are currently only known from Hong Kong, all are within one of the four IUCN threatened or near threatened categories listed. Seven species are recorded from Hong Kong for the first time. The moth assemblages recorded are typical of human disturbed forest, feng shui woods and orchards, with a relatively low Geometridae component, and includes a small number of species normally associated with agriculture and open habitats that were found in the SSSI site. Comparisons showed that each sub-site had a substantially different assemblage of species, thus the site as a whole should retain the mosaic of micro-habitats in order to maintain the high moth species richness observed. -

Suppression of Gene Juvenile Hormone Diol Kinase Delays Pupation in Heortia Vitessoides Moore
insects Article Suppression of Gene Juvenile Hormone Diol Kinase Delays Pupation in Heortia vitessoides Moore Zihao Lyu, Zhixing Li, Jie Cheng, Chunyan Wang, Jingxiang Chen and Tong Lin * College of Forestry and Landscape Architecture, South China Agricultural University, Guangzhou 510642, China * Correspondence: [email protected]; Tel.: +86-020-8528-2217 Received: 5 July 2019; Accepted: 25 August 2019; Published: 2 September 2019 Abstract: Juvenile hormone diol kinase (JHDK) is a critical enzyme involved in juvenile hormone degradation in insects. In this study, HvJHDK in the Heortia vitessoides Moore (Lepidoptera: Crambidae) transcriptional library was cloned. Stage-specific expression patterns of HvJHDK, HvJHEH, and HvJHE as well as juvenile hormone titers were determined. The three tested enzymes participated in juvenile hormone degradation. Moreover, juvenile hormone titers peaked after larval–larval molts, consistent with a role for juvenile hormone in inhibition of metamorphosis. HvJHDK was subsequently suppressed using RNA interference (RNAi) to reveal its functions. Different concentrations of dsJHDK elicited the optimal interference efficiency at different life stages of H. vitessoides. Suppression of HvJHDK decreased HvJHDK content and increased the juvenile hormone titer, thereby resulting in reduced triglyceride content, sharply declined survival rate, clearly lethal phenotypes, and extended larval growth. Moreover, suppression of HvJHDK upregulated HvJHEH and HvJHE expression levels, suggesting that there is feedback regulation in the juvenile hormone metabolic pathway. Taken together, our findings provide molecular references for the selection of novel insecticidal targets. Keywords: juvenile hormone diol kinase; juvenile hormone; Heortia vitessoides Moore; RNA interference; triglyceride 1. Introduction Juvenile hormone (JH) is one of the most important insect hormones. -

Butterflies and Moths of Gwinnett County, Georgia, United States
Heliothis ononis Flax Bollworm Moth Coptotriche aenea Blackberry Leafminer Argyresthia canadensis Apyrrothrix araxes Dull Firetip Phocides pigmalion Mangrove Skipper Phocides belus Belus Skipper Phocides palemon Guava Skipper Phocides urania Urania skipper Proteides mercurius Mercurial Skipper Epargyreus zestos Zestos Skipper Epargyreus clarus Silver-spotted Skipper Epargyreus spanna Hispaniolan Silverdrop Epargyreus exadeus Broken Silverdrop Polygonus leo Hammock Skipper Polygonus savigny Manuel's Skipper Chioides albofasciatus White-striped Longtail Chioides zilpa Zilpa Longtail Chioides ixion Hispaniolan Longtail Aguna asander Gold-spotted Aguna Aguna claxon Emerald Aguna Aguna metophis Tailed Aguna Typhedanus undulatus Mottled Longtail Typhedanus ampyx Gold-tufted Skipper Polythrix octomaculata Eight-spotted Longtail Polythrix mexicanus Mexican Longtail Polythrix asine Asine Longtail Polythrix caunus (Herrich-Schäffer, 1869) Zestusa dorus Short-tailed Skipper Codatractus carlos Carlos' Mottled-Skipper Codatractus alcaeus White-crescent Longtail Codatractus yucatanus Yucatan Mottled-Skipper Codatractus arizonensis Arizona Skipper Codatractus valeriana Valeriana Skipper Urbanus proteus Long-tailed Skipper Urbanus viterboana Bluish Longtail Urbanus belli Double-striped Longtail Urbanus pronus Pronus Longtail Urbanus esmeraldus Esmeralda Longtail Urbanus evona Turquoise Longtail Urbanus dorantes Dorantes Longtail Urbanus teleus Teleus Longtail Urbanus tanna Tanna Longtail Urbanus simplicius Plain Longtail Urbanus procne Brown Longtail -

Contributions to the Life-History of Tetraclinis Articu- Lata, Masters, with Some Notes on the Phylogeny of the Cupressoideae and Callitroideae
Contributions to the Life-history of Tetraclinis articu- lata, Masters, with some Notes on the Phylogeny of the Cupressoideae and Callitroideae. BY W. T. SAXTON, M.A., F.L.S., Professor of Botany at the Ahmedabad Institute of Science, India. With Plates XLIV-XLVI and nine Figures in the Text. INTRODUCTION. HE Gum Sandarach tree of Morocco and Algeria has been well known T to botanists from very early times. Some account of it is given by Hooker and Ball (20), who speak of the beauty and durability of the wood, and state that they consider the tree to be probably correctly identified with the Bvlov of the Odyssey (v. 60),1 and with the Ovlov and Ovia of Theo- phrastus (' Hist. PI.' v. 3, 7)/ as well as, undoubtedly, with the Citrus wood of the Romans. The largest trees met with by them, growing in an un- cultivated state, were about 30 feet high. The resin, known as sandarach, is stated to be collected by the Moors and exported to Europe, where it is used as a varnish. They quote Shaw (49 a and b) as having described and figured the tree under the name of Thuja articulata, in his ' Travels in Barbary'; this statement, however, is not accurate. In both editions of the work cited the plant is figured and described as ' Cupressus fructu quadri- valvi, foliis Equiseti instar articulatis '. Some interesting particulars of the use of the timber are given by Hansen (19), who also implies that the embryo has from three to six cotyledons. Both Hooker and Ball, and Hansen, followed by almost all others who have studied the plant, speak of it as Callitris qtiadrivalvis. -

Butterflies and Moths of Dorchester County, Maryland, United States
Heliothis ononis Flax Bollworm Moth Coptotriche aenea Blackberry Leafminer Argyresthia canadensis Apyrrothrix araxes Dull Firetip Phocides pigmalion Mangrove Skipper Phocides belus Belus Skipper Phocides palemon Guava Skipper Phocides urania Urania skipper Proteides mercurius Mercurial Skipper Epargyreus zestos Zestos Skipper Epargyreus clarus Silver-spotted Skipper Epargyreus spanna Hispaniolan Silverdrop Epargyreus exadeus Broken Silverdrop Polygonus leo Hammock Skipper Polygonus savigny Manuel's Skipper Chioides albofasciatus White-striped Longtail Chioides zilpa Zilpa Longtail Chioides ixion Hispaniolan Longtail Aguna asander Gold-spotted Aguna Aguna claxon Emerald Aguna Aguna metophis Tailed Aguna Typhedanus undulatus Mottled Longtail Typhedanus ampyx Gold-tufted Skipper Polythrix octomaculata Eight-spotted Longtail Polythrix mexicanus Mexican Longtail Polythrix asine Asine Longtail Polythrix caunus (Herrich-Schäffer, 1869) Zestusa dorus Short-tailed Skipper Codatractus carlos Carlos' Mottled-Skipper Codatractus alcaeus White-crescent Longtail Codatractus yucatanus Yucatan Mottled-Skipper Codatractus arizonensis Arizona Skipper Codatractus valeriana Valeriana Skipper Urbanus proteus Long-tailed Skipper Urbanus viterboana Bluish Longtail Urbanus belli Double-striped Longtail Urbanus pronus Pronus Longtail Urbanus esmeraldus Esmeralda Longtail Urbanus evona Turquoise Longtail Urbanus dorantes Dorantes Longtail Urbanus teleus Teleus Longtail Urbanus tanna Tanna Longtail Urbanus simplicius Plain Longtail Urbanus procne Brown Longtail -

Hawaiian Hoary Bat Habitat
Flexible Foraging of the Hawaiian Hoary Bat on Maui An update of the H. T. Harvey & Associates ecological research on Opeapea Dave Johnston, Kristin Jonasson, and Brad Yuen ESRC Meeting Honolulu, Hawaii 5 March 2020 Strategy for Recovery of the Species • Which habitats do they use? • How much land does a bat use? • What do they eat? Study Area & Bat Detector Locations General Random Tessellation Stratified survey design Acoustic Monitoring - Methods Bi-monthly 9 habitats 3 nights 5 replicates / month 315 total deployments SM4 bat detectors 35 bat detector sites per each habitat Relationship between Precipitation and the Nine Habitats in the Study Area. Modelling of Acoustic Data - Methods • Differences between habitat activity - generalized linear model fit by maximum likelihood with a negative binomial distribution date and site as random factors, and habitat as fixed effect of interest. • Tested for differences between months within each habitat, habitats within each month, using pairwise contrasts with a Tukey adjustment for comparing among estimates Methods: Determining Core Use Areas Mist-netting at “hot spots” of bat activity Radio-telemetry of foraging bats using triangulation by mobile antennae and fixed stations Analysis of data to determine core use area (CUA) 50% Kernel and foraging range (FR) 95% kernel. Reanalized data using methods described in Bonaccorso et al. 2015. Prey Availability Bi-monthly UV light collection trap 9 habitats Extraordinary sample set Identification of Moths from Light Traps Moths ID by Matt Medeiros Slide preparations of Male Genitalia of Darna pallivitta and Macaria abydata Used to Identify the Species. Modelling of Dry Weights of Insects per Habitat and Month • We fit a negative binomial generalized linear model with a log link function from the MASS package (Venables and Ripley 2002) • We used the estimated marginal means (emmeans) (Searle et al. -

Seiridium Canker of Cypress Trees in Arizona Jeff Schalau
ARIZONA COOPERATIVE E TENSION AZ1557 January 2012 Seiridium Canker of Cypress Trees in Arizona Jeff Schalau Introduction Leyland cypress (x Cupressocyparis leylandii) is a fast- growing evergreen that has been widely planted as a landscape specimen and along boundaries to create windbreaks or privacy screening in Arizona. The presence of Seiridium canker was confirmed in Prescott, Arizona in July 2011 and it is suspected that the disease occurs in other areas of the state. Seiridium canker was first identified in California’s San Joaquin Valley in 1928. Today, it can be found in Europe, Asia, New Zealand, Australia, South America and Africa on plants in the cypress family (Cupressaceae). Leyland cypress, Monterey cypress, (Cupressus macrocarpa) and Italian cypress (C. sempervirens) are highly susceptible and can be severely impacted by this disease. Since Leyland and Italian cypress have been widely planted in Arizona, it is imperative that Seiridium canker management strategies be applied and suitable resistant tree species be recommended for planting in the future. The Pathogen Seiridium canker is known to be caused by three different fungal species: Seiridium cardinale, S. cupressi and S. unicorne. S. cardinale is the most damaging of the three species and is SCHALAU found in California. S. unicorne and S. cupressi are found in the southeastern United States where the primary host is JEFF Leyland cypress. All three species produce asexual fruiting Figure 1. Leyland cypress tree with dead branch (upper left) and main leader bodies (acervuli) in cankers. The acervuli produce spores caused by Seiridium canker. (conidia) which spread by water, human activity (pruning and transport of infected plant material), and potentially insects, birds and animals to neighboring trees where new Symptoms and Signs infections can occur. -

Development, Plasticity and Evolution of Butterfly Eyespot Patterns" (1996), by Paul M
Published on The Embryo Project Encyclopedia (https://embryo.asu.edu) "Development, Plasticity and Evolution of Butterfly Eyespot Patterns" (1996), by Paul M. Brakefield et al. [1] By: Zou, Yawen Keywords: Butterfly eyespots [2] Paul M. Brakefield experiment [3] Bush-brown butterfly [4] eyespot patterns [5] Paul M. Brakefield and his research team in Leiden, the Netherlands, examined the development, plasticity, ande volution [6] of butterfly [7] eyespot patterns, and published their findings in Nature in 1996. Eyespots are eye-shaped color patterns that appear on the wings of some butterflies and birds [8] as well as on the skin of some fish [9] and reptiles. In butterflies, such as the peacock butterfly [7] (Aglais io [10]), the eyespots resemble the eyes of birds [8] and help butterflies deter potential predators. Brakefield's research team described the stages through which eyespots develop, identified the genes [11] and environmental signals that affect eye-spot appearance in some species, and demonstrated that small genetic variations can change butterfly [7] eyespot color and shape. The research focused on a few butterfly [7] species, but it contributed to more general claims of how the environment may affect the development of coloration and how specific color patterns may have evolved. At the time of experiment, Brakefield was a Chair in Evolutionary Biology at the University of Leiden [12], in Leiden, the Netherlands, though in 2010 he moved to Cambridge, UK to direct the University Museum of Zoology. While in Leiden, he persued a research program in evolutionary developmental biology [13] and had started working on butterfly [7] eyespots in collaboration with Vernon French, who was at the University of Edinburgh [14] in Edinburgh, Scotland. -

Title Production Study of the Population of the Pine Caterpillar
Production Study of the Population of the Pine Caterpillar Title (Dendrolimus spectabilis Butler) Author(s) Kikuzawa, Kihachiro; Furuno, Tooshu Citation 京都大学農学部演習林報告 (1971), 42: 16-26 Issue Date 1971-03-25 URL http://hdl.handle.net/2433/191500 Right Type Departmental Bulletin Paper Textversion publisher Kyoto University 16 Production Study of the Population of the Pine Caterpillar (Dendrolimus spectabilis Butler) Kihachiro KIKUZAWA* and Tooshu FURUNO** マツカ レハ幼虫個体群の生物生産の研究 菊 沢 喜 八 郎*・ 古 野 東 洲** CONTENTS Résumé 16 I . Metabolism of Individual Larva V Pi 17 II. Estimation of population Density Introduction 17 III. production of population Material And Method 18 Discussion 24 Results 19 Literature 26 RESUME Productivity of a pine caterpillar population was investigated in the present study. The changes in larval density of the pine caterpillar population were estimated by the fecal-pellets counting method and by the direct counting method at a pine stand in a nursery. The food consumption, assimilation, respiration and growth were also mea- sured in the laboratory. The total food consumption, assimilation, respiration and growth of the population were calculated utilizing the individual values and population density. An individual larva consumed about 14.0g of pine needles over one growing season. From which 11.2g was defecated as feces, and the rest 2.8g was assimilated. 2.3g, or eighty per cent of the assimilated matter was used for respiration and 0.5g was used for growth of larval body weight. Larval density was estimated at about 63 individuals per square meter in September 1967, which decreased to about 6 individuals per square meter in July 1968 (at the time of pupation). -

Studies on the Biology and Ecology
STUDIES ON THE BIOLOGY AND ECOLOGY OF THE CABBAGE MOTH, MAMESTRA BRASSICAE L. (LEPIDOPTERA : NOCTUIDAE) by AQUILES MONTAGNE Ingeniero Agronomo (Venezuela) A thesis submitted for the degree of Doctor of Philosophy of the University of London and the Diploma of Imperial College Department of Zoology and Applied Entomology, Imperial College Field Station, Silwood Park, Ascot Berkshire September 1977 TABLE OF CONTENT Page ABSTRACT ' GENERAL INTRODUCTION 2 SECTION 1 THE BIOLOGY OF M. brassicae 5 1.1 Introduction 5 1.2 General Description of the Stages 6 1.3 Life History and Habits 8 1.4 Laboratory Studies on the Effect of Temperature on the Development and Survival of the Immature Stages 25 1.4.1 Materials and methods 25 1.4.2 Results and discussion 26 1.5 Laboratory Studies on Longevity and Fecundity 36 1.5.1 Materials and methods 36 1.5.2 Results and discussion 37 1.5.2.1 Longevity 37 1.5.2.2 Fecundity and Fertility 38 ' 1.6 Section General Discussion 47 SECTION 2 STUDIES ON THE EFFECTS OF LARVAL DENSITY ON M. 50 brassicae 2.1 Introduction 50 2.2 Review of Literature 50 2.3 Material and Methods 59 ii Table of Contents (Continued) Page 2.4 Results and Discussion 60 2.4.1 Colour variations in larval stage 60 2.4.1.1 Larval colour types 60 2.4.1.2 The colour of larvae reared at various densities 62 2.4.2 The pattern of larval and pupal development 66 2.4.2.1 The duration of larval development 66 2.4.2.2 The pattern of larval growth 71 2.4.2.3 The duration of prepupal and pupal periods 72 2.4.2.4 Larval and pupal mortality 75 2.4.2.5 Sex ratio -

Influence of Developmental Conditions on Immune Function and Dispersal-Related Traits in the Glanville Fritillary (Melitaea Cinxia) Butterfly
Influence of Developmental Conditions on Immune Function and Dispersal-Related Traits in the Glanville Fritillary (Melitaea cinxia) Butterfly Marjo Saastamoinen1*, Markus J. Rantala2 1 Department of Biological Sciences, University of Helsinki, Helsinki, Finland, 2 Department of Biology, University of Turku, Turku, Finland Abstract Organisms in the wild are constantly faced with a wide range of environmental variability, such as fluctuation in food availability. Poor nutritional conditions influence life-histories via individual resource allocation patterns, and trade-offs between competing traits. In this study, we assessed the influence of food restriction during development on the energetically expensive traits flight metabolic rate (proxy of dispersal ability), encapsulation rate (proxy of immune defence), and lifespan using the Glanville fritillary butterfly, Melitaea cinxia, as a model organism. Additionally, we examined the direct costs of flight on individual immune function, and whether those costs increase under restricted environmental conditions. We found that nutritional restriction during development enhanced adult encapsulations rate, but reduced both resting and flight metabolic rates. However, at the individual level metabolic rates were not associated with encapsulation rate. Interestingly, individuals that were forced to fly prior to the immune assays had higher encapsulation rates than individuals that had not flown, suggesting that flying itself enhances immune response. Finally, in the control group encapsulation rate correlated positively with lifespan, whereas in the nutritional restriction group there was no relationship between these traits, suggesting that the association between encapsulation rate on adult lifespan was condition-dependent. Thus stressful events during both larval development (food limitation) and adulthood (forced flight) induce increased immune response in the adult butterflies, which may allow individuals to cope with stressful events later on in life. -
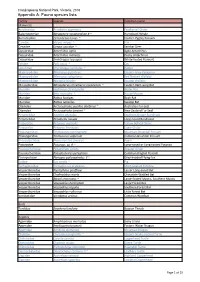
Report-VIC-Croajingolong National Park-Appendix A
Croajingolong National Park, Victoria, 2016 Appendix A: Fauna species lists Family Species Common name Mammals Acrobatidae Acrobates pygmaeus Feathertail Glider Balaenopteriae Megaptera novaeangliae # ~ Humpback Whale Burramyidae Cercartetus nanus ~ Eastern Pygmy Possum Canidae Vulpes vulpes ^ Fox Cervidae Cervus unicolor ^ Sambar Deer Dasyuridae Antechinus agilis Agile Antechinus Dasyuridae Antechinus mimetes Dusky Antechinus Dasyuridae Sminthopsis leucopus White-footed Dunnart Felidae Felis catus ^ Cat Leporidae Oryctolagus cuniculus ^ Rabbit Macropodidae Macropus giganteus Eastern Grey Kangaroo Macropodidae Macropus rufogriseus Red Necked Wallaby Macropodidae Wallabia bicolor Swamp Wallaby Miniopteridae Miniopterus schreibersii oceanensis ~ Eastern Bent-wing Bat Muridae Hydromys chrysogaster Water Rat Muridae Mus musculus ^ House Mouse Muridae Rattus fuscipes Bush Rat Muridae Rattus lutreolus Swamp Rat Otariidae Arctocephalus pusillus doriferus ~ Australian Fur-seal Otariidae Arctocephalus forsteri ~ New Zealand Fur Seal Peramelidae Isoodon obesulus Southern Brown Bandicoot Peramelidae Perameles nasuta Long-nosed Bandicoot Petauridae Petaurus australis Yellow Bellied Glider Petauridae Petaurus breviceps Sugar Glider Phalangeridae Trichosurus cunninghami Mountain Brushtail Possum Phalangeridae Trichosurus vulpecula Common Brushtail Possum Phascolarctidae Phascolarctos cinereus Koala Potoroidae Potorous sp. # ~ Long-nosed or Long-footed Potoroo Pseudocheiridae Petauroides volans Greater Glider Pseudocheiridae Pseudocheirus peregrinus