Differential Reactivity of Microglia in Two Mouse Models of Multiple Sclerosis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

WO 2016/147053 Al 22 September 2016 (22.09.2016) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2016/147053 Al 22 September 2016 (22.09.2016) P O P C T (51) International Patent Classification: (71) Applicant: RESVERLOGIX CORP. [CA/CA]; 300, A61K 31/551 (2006.01) A61P 37/02 (2006.01) 4820 Richard Road Sw, Calgary, AB, T3E 6L1 (CA). A61K 31/517 (2006.01) C07D 239/91 (2006.01) (72) Inventors: WASIAK, Sylwia; 431 Whispering Water (21) International Application Number: Trail, Calgary, AB, T3Z 3V1 (CA). KULIKOWSKI, PCT/IB20 16/000443 Ewelina, B.; 31100 Swift Creek Terrace, Calgary, AB, T3Z 0B7 (CA). HALLIDAY, Christopher, R.A.; 403 (22) International Filing Date: 138-18th Avenue SE, Calgary, AB, T2G 5P9 (CA). GIL- 10 March 2016 (10.03.2016) HAM, Dean; 249 Scenic View Close NW, Calgary, AB, (25) Filing Language: English T3L 1Y5 (CA). (26) Publication Language: English (81) Designated States (unless otherwise indicated, for every kind of national protection available): AE, AG, AL, AM, (30) Priority Data: AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, 62/132,572 13 March 2015 (13.03.2015) US BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, 62/264,768 8 December 2015 (08. 12.2015) US DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, [Continued on nextpage] (54) Title: COMPOSITIONS AND THERAPEUTIC METHODS FOR THE TREATMENT OF COMPLEMENT-ASSOCIATED DISEASES (57) Abstract: The invention comprises methods of modulating the complement cascade in a mammal and for treating and/or preventing diseases and disorders as sociated with the complement pathway by administering a compound of Formula I or Formula II, such as, for example, 2-(4-(2-hydroxyethoxy)-3,5-dimethylphenyl)- 5,7-dimethoxyquinazolin-4(3H)-one or a pharmaceutically acceptable salt thereof. -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -
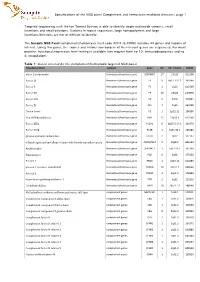
Specifications of the NGS Panel Complement and Hemostasis Mediated Diseases| Page 1
Specifications of the NGS panel Complement and hemostasis mediated diseases| page 1 Targeted sequencing with the Ion Torrent System is able to identify single nucleotide variants, small insertions and small deletions. Variants in repeat sequences, large homopolymers and large insertions/deletions are not or difficult to identify. The Sanquin NGS Panel complement/hemostasis (test code X001 to X006) includes 44 genes and regions of interest. Using this panel, the exones and intron/exon borders of the relevant genes are sequenced. For most proteins, functional/expression level testing is available (see request form no 10: immunodiagnostics and no 4: coagulation). Table 1: Genes covered by the complement/hemostasis targeted NGS panel. Encoded protein Context Gene Chr Chr. Positie OMIM alpha 2-antiplasmin Hemostasis/trombosis gene SERPINF2 17 17p13 613168 Factor IX Hemostasis/trombosis gene F9 X Xq27.1-27.2 300746 Factor V Hemostasis/trombosis gene F5 1 1q23 612309 Factor VII Hemostasis/trombosis gene F7 13 13q34 613878 Factor VIII Hemostasis/trombosis gene F8 X Xq28 300841 Factor XI Hemostasis/trombosis gene F11 4 4q35 264900 Tissue factor Hemostasis/trombosis gene F3 1 1p22-21 134390 Von Willebrand factor Hemostasis/trombosis gene VWF 12 12p13.3 613160 Factor XIIIa Hemostasis/trombosis gene F13A1 6 6p25.3-24.3 134570 Factor XIIIb Hemostasis/trombosis gene F13B 1 1q31-32.1 134580 gamma-glutamyl carboxylase Hemostasis/trombosis gene GGCX 2 2p12 137167 a Disintergrin and metalloproteinase wih thrombospondin repeats Hemostasis/trombosis gene -

A Case of Meningococcal Sepsis and Meningitis with Complement 7
Case Report Infection & http://dx.doi.org/10.3947/ic.2013.45.1.94 Infect Chemother 2013;45(1):94-98 Chemotherapy pISSN 2093-2340 · eISSN 2092-6448 A Case of Meningococcal Sepsis and Meningitis with Complement 7 Deficiency in a Military Trainee Sung Hoon Sim1*, Jung Yeon Heo2*, Eui-Chong Kim3, and Kang-Won Choe2 1Department of Internal Medicine, Seoul National University Hospital, Seoul; 2Department of Internal Medicine, Armed Forces Capital Hospital, Seongnam; 3Department of Laboratory Medicine, Seoul National University Hospital, Seoul, Korea Complement component 7 (C7) deficiency leads to the loss of complement lytic function, and affected patients show in- creased susceptibility to encapsulated organisms infection, especially Neisseria meningitidis. Recently, we have experi- enced a 20-year-old military trainee with meningococcal sepsis and meningitis who was diagnosed as having C7 deficiency based upon the undetectable serum C7 protein on radioimmunoassay. This case emphasizes that although C7 deficiency is rare immune disorder, it is important to be aware of possibility about late complement deficiency among patients who pres- ent with meningococcal disease. Key Words: Neisseria meningitidis, Complement 7 deficiency, Meningitis, Sepsis Introduction of 20 and college freshmen residing in the dormitories are a well-known high risk group. The incidence of meningococcal Humans are the only host for Neisseria meningitidis which disease increases as those high risk age groups from different colonizes the nasopharynx and it is transmitted from human geological areas start communal living [2]. to human by infected respiratory secretions or saliva via air- Apart from these environmental factors, host factors such as borne respiratory droplets. -

A Computational Model for the Evaluation of Complement System Regulation Under Homeostasis, Disease, and Drug Intervention
bioRxiv preprint doi: https://doi.org/10.1101/225029; this version posted November 25, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. A Computational Model for the Evaluation of Complement System Regulation under Homeostasis, Disease, and Drug Intervention Nehemiah Zewde and Dimitrios Morikis* Department of Bioengineering University of California, Riverside *Corresponding author. E-mail address: [email protected] (D. Morikis) Key words: Computational ODE model; immune system regulation; complement system pathways; disease model; Factor H; compstatin; eculizumab. Page 1 of 33 bioRxiv preprint doi: https://doi.org/10.1101/225029; this version posted November 25, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Highlights • Computational model describing dynamics of complement system activation pathways • Complement dysregulation leads to deviation from homeostasis and to inflammatory diseases • Model identifies biomarkers to quantify the effects of complement dysregulation • Known drugs restore impaired dynamics of complement biomarkers under dysregulation • Disease-specific models are suitable for diagnosis and patient-specific drug treatment Page 2 of 33 bioRxiv preprint doi: https://doi.org/10.1101/225029; this version posted November 25, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract The complement system is a part of innate immunity that rapidly removes invading pathogens and impaired host-cells. Activation of the complement system is balanced under homeostasis by regulators that protect healthy host-cells. -

(MDR/TAP), Member 1 ABL1 NM 00
Official Symbol Accession Official Full Name ABCB1 NM_000927.3 ATP-binding cassette, sub-family B (MDR/TAP), member 1 ABL1 NM_005157.3 c-abl oncogene 1, non-receptor tyrosine kinase ADA NM_000022.2 adenosine deaminase AHR NM_001621.3 aryl hydrocarbon receptor AICDA NM_020661.1 activation-induced cytidine deaminase AIRE NM_000383.2 autoimmune regulator APP NM_000484.3 amyloid beta (A4) precursor protein ARG1 NM_000045.2 arginase, liver ARG2 NM_001172.3 arginase, type II ARHGDIB NM_001175.4 Rho GDP dissociation inhibitor (GDI) beta ATG10 NM_001131028.1 ATG10 autophagy related 10 homolog (S. cerevisiae) ATG12 NM_004707.2 ATG12 autophagy related 12 homolog (S. cerevisiae) ATG16L1 NM_198890.2 ATG16 autophagy related 16-like 1 (S. cerevisiae) ATG5 NM_004849.2 ATG5 autophagy related 5 homolog (S. cerevisiae) ATG7 NM_001136031.2 ATG7 autophagy related 7 homolog (S. cerevisiae) ATM NM_000051.3 ataxia telangiectasia mutated B2M NM_004048.2 beta-2-microglobulin B3GAT1 NM_018644.3 beta-1,3-glucuronyltransferase 1 (glucuronosyltransferase P) BATF NM_006399.3 basic leucine zipper transcription factor, ATF-like BATF3 NM_018664.2 basic leucine zipper transcription factor, ATF-like 3 BAX NM_138761.3 BCL2-associated X protein BCAP31 NM_005745.7 B-cell receptor-associated protein 31 BCL10 NM_003921.2 B-cell CLL/lymphoma 10 BCL2 NM_000657.2 B-cell CLL/lymphoma 2 BCL2L11 NM_138621.4 BCL2-like 11 (apoptosis facilitator) BCL3 NM_005178.2 B-cell CLL/lymphoma 3 BCL6 NM_001706.2 B-cell CLL/lymphoma 6 BID NM_001196.2 BH3 interacting domain death agonist BLNK NM_013314.2 -

Insights Dust Mite by Influenza a Infection
Downloaded from http://www.jimmunol.org/ by guest on September 29, 2021 is online at: average * The Journal of Immunology , 10 of which you can access for free at: 2012; 188:832-843; Prepublished online 14 from submission to initial decision 4 weeks from acceptance to publication December 2011; doi: 10.4049/jimmunol.1102349 http://www.jimmunol.org/content/188/2/832 Shifting of Immune Responsiveness to House Dust Mite by Influenza A Infection: Genomic Insights Amal Al-Garawi, Mainul Husain, Dora Ilieva, Alison A. Humbles, Roland Kolbeck, Martin R. Stampfli, Paul M. O'Byrne, Anthony J. Coyle and Manel Jordana J Immunol cites 38 articles Submit online. Every submission reviewed by practicing scientists ? is published twice each month by Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://jimmunol.org/subscription http://www.jimmunol.org/content/suppl/2011/12/14/jimmunol.110234 9.DC1 This article http://www.jimmunol.org/content/188/2/832.full#ref-list-1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material References Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2012 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of September 29, 2021. The Journal of Immunology Shifting of Immune Responsiveness to House Dust Mite by Influenza A Infection: Genomic Insights Amal Al-Garawi,*,†,1 Mainul Husain,*,†,1 Dora Ilieva,*,† Alison A. -

Supplement, Table 8. Genes Affected by Celecoxib in "Immune Response" Gene Ontology Category
Supplement, Table 8. Genes affected by Celecoxib in "immune response" Gene Ontology category Geometri UniGene HUGO Parametric Gene c mean of Cluster ID Symbol p-value ratios P/F Hs.25647 FOS v-fos FBJ murine osteosarcoma viral oncogene homolog 0.621 0.0407 Hs.423 PAP pancreatitis-associated protein 0.683 0.0040 Hs.407546 TNFAIP6 tumor necrosis factor, alpha-induced protein 6 0.736 0.0053 Hs.308680 C1R complement component 1, r subcomponent 0.751 0.0179 Hs.789 CXCL1 chemokine (C-X-C motif) ligand 1 (melanoma growth stimulating activity, alpha) 0.762 0.0579 Hs.110675 APOE apolipoprotein E 0.762 0.0253 Hs.407506 INHA inhibin, alpha 0.764 0.0185 Hs.78065 C7 complement component 7 0.765 0.0081 Hs.134231 VPS45A vacuolar protein sorting 45A (yeast) 0.78 0.0000 Hs.436042 CXCL12 chemokine (C-X-C motif) ligand 12 (stromal cell-derived factor 1) 0.788 0.0189 Hs.172674 NFATC3 nuclear factor of activated T-cells, cytoplasmic, calcineurin-dependent 3 0.793 0.0019 Hs.421342 STAT3 signal transducer and activator of transcription 3 (acute-phase response factor) 0.799 0.0314 Hs.1735 INHBB inhibin, beta B (activin AB beta polypeptide) 0.801 0.0311 Hs.458355 C1S complement component 1, s subcomponent 0.815 0.0238 Hs.77546 ANKRD15 ankyrin repeat domain 15 0.818 0.0017 GALNAC4S- Hs.523379 6ST B cell RAG associated protein 0.829 0.0522 Hs.421194 TPST1 tyrosylprotein sulfotransferase 1 0.831 0.0206 Hs.7957 ADAR adenosine deaminase, RNA-specific 0.834 0.0210 Hs.374357 NFRKB nuclear factor related to kappa B binding protein 0.849 0.0104 Hs.1281 C5 complement component -
Table 2: Complete List of Genes That Were Down-Regulated (> 3-Fold) at Week 6 with Dynamic Compressive Loading Compared to CM+ Free-Swelling Control
Table 2: Complete list of genes that were down-regulated (> 3-fold) at week 6 with dynamic compressive loading compared to CM+ free-swelling control. Probe Set ID Gene Symbol Gene Title Bt.9807.2.S1_at GPNMB glycoprotein (transmembrane) nmb Bt.9675.1.S1_at LOC787253 similar to Extracellular proteinase inhibitor Bt.9351.2.S1_at MGC139990 similar to EVIN1 Bt.9082.1.S1_at LGALS4 lectin, galactoside-binding, soluble, 4 (galectin 4) Bt.8421.2.S1_at --- Transcribed locus, strongly similar to NP_001002374.1 hypothetical protein LOC43 Bt.8328.1.S1_at LOC521189 similar to NRAMP2 Bt.7210.1.S1_at PRND prion protein 2 (dublet) Bt.6392.1.S1_at LOC527597 similar to transient receptor potential cation channel subfamily M member 6 Bt.5373.1.S1_at SLC11A1 solute carrier family 11 (proton-coupled divalent metal ion transporters), membe Bt.502.1.A1_at RGN regucalcin (senescence marker protein-30) Bt.4781.1.S1_at ADAM10 ADAM metallopeptidase domain 10 Bt.4604.1.S1_a_at BUCS1 butyryl Coenzyme A synthetase 1 Bt.422.1.S2_at IGFBP3 insulin-like growth factor binding protein 3 Bt.390.1.S1_at CALB3 calbindin 3, (vitamin D-dependent calcium binding protein) Bt.361.1.S1_at SLC1A3 solute carrier family 1 (glial high affinity glutamate transporter), member 3 Bt.352.1.S1_at PTGFR prostaglandin F receptor (FP) Bt.3189.1.A1_at LOC540831 similar to FLJ20273 protein Bt.29882.1.S1_at MITF microphthalmia-associated transcription factor Bt.29718.1.S1_a_at GHR growth hormone receptor Bt.28685.1.S1_at LOC540831 similar to FLJ20273 protein Bt.2846.1.A1_at MGC154996 similar to -

Characterising the Role of CR1 and CR2 in a Humanised Mouse Model
Characterising the Role of CR1 and CR2 in a Humanised Mouse Model Harriet Michaela Williams Thesis presented for the degree of MPhil 2016 Division of Infection and Immunity School of Medicine Cardiff University Heath Park Cardiff, CF14 4XN DECLARATION This work has not been submitted in substance for any other degree or award at this or any other university or place of learning, nor is being submitted concurrently in candidature for any degree or other award. Signed (Harriet Williams) Date: 24 Oct 2016 STATEMENT 1 This thesis is being submitted in partial fulfillment of the requirements for the degree of MPhil Signed (Harriet Williams) Date: 24 Oct 2016 STATEMENT 2 This thesis is the result of my own independent work/investigation, except where otherwise stated, and the thesis has not been edited by a third party beyond what is permitted by Cardiff University’s Policy on the Use of Third Party Editors by Research Degree Students. Other sources are acknowledged by explicit references. The views expressed are my own. Signed (Harriet Williams) Date: 24 Oct 2016 STATEMENT 3 I hereby give consent for my thesis, if accepted, to be available online in the University’s Open Access repository and for inter-library loan, and for the title and summary to be made available to outside organisations. Signed (Harriet Williams) Date: 24 Oct 2016 STATEMENT 4: PREVIOUSLY APPROVED BAR ON ACCESS I hereby give consent for my thesis, if accepted, to be available online in the University’s Open Access repository and for inter-library loans after expiry of a bar on access previously approved by the Academic Standards & Quality Committee. -

Proteomic Landscape of the Human Choroid–Retinal Pigment Epithelial Complex
Supplementary Online Content Skeie JM, Mahajan VB. Proteomic landscape of the human choroid–retinal pigment epithelial complex. JAMA Ophthalmol. Published online July 24, 2014. doi:10.1001/jamaophthalmol.2014.2065. eFigure 1. Choroid–retinal pigment epithelial (RPE) proteomic analysis pipeline. eFigure 2. Gene ontology (GO) distributions and pathway analysis of human choroid– retinal pigment epithelial (RPE) protein show tissue similarity. eMethods. Tissue collection, mass spectrometry, and analysis. eTable 1. Complete table of proteins identified in the human choroid‐RPE using LC‐ MS/MS. eTable 2. Top 50 signaling pathways in the human choroid‐RPE using MetaCore. eTable 3. Top 50 differentially expressed signaling pathways in the human choroid‐RPE using MetaCore. eTable 4. Differentially expressed proteins in the fovea, macula, and periphery of the human choroid‐RPE. eTable 5. Differentially expressed transcription proteins were identified in foveal, macular, and peripheral choroid‐RPE (p<0.05). eTable 6. Complement proteins identified in the human choroid‐RPE. eTable 7. Proteins associated with age related macular degeneration (AMD). This supplementary material has been provided by the authors to give readers additional information about their work. © 2014 American Medical Association. All rights reserved. 1 Downloaded From: https://jamanetwork.com/ on 09/25/2021 eFigure 1. Choroid–retinal pigment epithelial (RPE) proteomic analysis pipeline. A. The human choroid‐RPE was dissected into fovea, macula, and periphery samples. B. Fractions of proteins were isolated and digested. C. The peptide fragments were analyzed using multi‐dimensional LC‐MS/MS. D. X!Hunter, X!!Tandem, and OMSSA were used for peptide fragment identification. E. Proteins were further analyzed using bioinformatics. -

Genome-Wide Expression Differences in Anti-Vegf and Dexamethasone
www.nature.com/scientificreports OPEN Genome-wide expression differences in anti-Vegf and dexamethasone treatment of Received: 10 November 2016 Accepted: 22 June 2017 inflammatory angiogenesis in the Published: xx xx xxxx rat cornea Pierfrancesco Mirabelli, Anthony Mukwaya , Anton Lennikov, Maria Xeroudaki, Beatrice Peebo, Mira Schaupper & Neil Lagali Angiogenesis as a pathological process in the eye can lead to blindness. In the cornea, suppression of angiogenesis by anti-VEGF treatment is only partially effective while steroids, although effective in treating inflammation and angiogenesis, have broad activity leading to undesirable side effects. In this study, genome-wide expression was investigated in a suture-induced corneal neovascularization model in rats, to investigate factors differentially targeted by dexamethasone and anti-Vegf. Topical treatment with either rat-specific anti-Vegf, dexamethasone, or normal goat IgG (sham) was given to sutured corneas for 48 hours, after which in vivo imaging, tissue processing for RNA microarray, and immunofluorescence were performed. Dexamethasone suppressed limbal vasodilation (P < 0.01) and genes in PI3K-Akt, focal adhesion, and chemokine signaling pathways more effectively than anti-Vegf. The most differentially expressed genes were confirmed by immunofluorescence, qRTPCR and Western blot. Strong suppression of Reg3g and the inflammatory chemokinesCcl2 and Cxcl5 and activation of classical complement pathway factors C1r, C1s, C2, and C3 occurred with dexamethasone treatment, effects absent with anti-Vegf treatment. The genome-wide results obtained in this study provide numerous potential targets for specific blockade of inflammation and angiogenesis in the cornea not addressed by anti-Vegf treatment, as possible alternatives to broad-acting immunosuppressive therapy. Angiogenesis is an essential physiologic process occurring during embryogenesis as well as in adult organisms in the context of wound healing, muscle growth, and the menstrual cycle.