Characterising the Role of CR1 and CR2 in a Humanised Mouse Model
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

WO 2016/147053 Al 22 September 2016 (22.09.2016) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2016/147053 Al 22 September 2016 (22.09.2016) P O P C T (51) International Patent Classification: (71) Applicant: RESVERLOGIX CORP. [CA/CA]; 300, A61K 31/551 (2006.01) A61P 37/02 (2006.01) 4820 Richard Road Sw, Calgary, AB, T3E 6L1 (CA). A61K 31/517 (2006.01) C07D 239/91 (2006.01) (72) Inventors: WASIAK, Sylwia; 431 Whispering Water (21) International Application Number: Trail, Calgary, AB, T3Z 3V1 (CA). KULIKOWSKI, PCT/IB20 16/000443 Ewelina, B.; 31100 Swift Creek Terrace, Calgary, AB, T3Z 0B7 (CA). HALLIDAY, Christopher, R.A.; 403 (22) International Filing Date: 138-18th Avenue SE, Calgary, AB, T2G 5P9 (CA). GIL- 10 March 2016 (10.03.2016) HAM, Dean; 249 Scenic View Close NW, Calgary, AB, (25) Filing Language: English T3L 1Y5 (CA). (26) Publication Language: English (81) Designated States (unless otherwise indicated, for every kind of national protection available): AE, AG, AL, AM, (30) Priority Data: AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, 62/132,572 13 March 2015 (13.03.2015) US BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, 62/264,768 8 December 2015 (08. 12.2015) US DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, [Continued on nextpage] (54) Title: COMPOSITIONS AND THERAPEUTIC METHODS FOR THE TREATMENT OF COMPLEMENT-ASSOCIATED DISEASES (57) Abstract: The invention comprises methods of modulating the complement cascade in a mammal and for treating and/or preventing diseases and disorders as sociated with the complement pathway by administering a compound of Formula I or Formula II, such as, for example, 2-(4-(2-hydroxyethoxy)-3,5-dimethylphenyl)- 5,7-dimethoxyquinazolin-4(3H)-one or a pharmaceutically acceptable salt thereof. -

Expressed Sequence Tag Analysis of the Response of Apple
Physiologia Plantarum 133: 298–317. 2008 Copyright ª Physiologia Plantarum 2008, ISSN 0031-9317 Expressed sequence tag analysis of the response of apple (Malus x domestica ‘Royal Gala’) to low temperature and water deficit Michael Wisniewskia,*, Carole Bassetta,*, John Norellia, Dumitru Macarisina, Timothy Artlipa, Ksenija Gasicb and Schuyler Korbanb aUnited States Department of Agriculture – Agricultural Research Service (USDA-ARS), Appalachian Fruit Research Station, 2217 Wiltshire Road, Kearneysville, WV 25430, USA bDepartment of Natural Resources and Environmental Sciences, University of Illinois, 310 ERML, 1201 W. Gregory Drive, Urbana, IL 61801, USA Correspondence Leaf, bark, xylem and root tissues were used to make nine cDNA libraries from *Corresponding author, non-stressed (control) ‘Royal Gala’ apple trees, and from ‘Royal Gala’ trees e-mail: [email protected]; exposed to either low temperature (5°C for 24 h) or water deficit (45% of [email protected] saturated pot mass for 2 weeks). Over 22 600 clones from the nine libraries # Received 26 September 2007; revised 3 were subjected to 5 single-pass sequencing, clustered and annotated using January 2008 BLASTX. The number of clusters in the libraries ranged from 170 to 1430. Regarding annotation of the sequences, BLASTX analysis indicated that within doi: 10.1111/j.1399-3054.2008.01063.x the libraries 65–72% of the clones had a high similarity to known function genes, 6–15% had no functional assignment and 15–26% were completely novel. The expressed sequence tags were combined into three classes (control, low-temperature and water deficit) and the annotated genes in each class were placed into 1 of 10 different functional categories. -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

The Genetics of Bipolar Disorder
Molecular Psychiatry (2008) 13, 742–771 & 2008 Nature Publishing Group All rights reserved 1359-4184/08 $30.00 www.nature.com/mp FEATURE REVIEW The genetics of bipolar disorder: genome ‘hot regions,’ genes, new potential candidates and future directions A Serretti and L Mandelli Institute of Psychiatry, University of Bologna, Bologna, Italy Bipolar disorder (BP) is a complex disorder caused by a number of liability genes interacting with the environment. In recent years, a large number of linkage and association studies have been conducted producing an extremely large number of findings often not replicated or partially replicated. Further, results from linkage and association studies are not always easily comparable. Unfortunately, at present a comprehensive coverage of available evidence is still lacking. In the present paper, we summarized results obtained from both linkage and association studies in BP. Further, we indicated new potential interesting genes, located in genome ‘hot regions’ for BP and being expressed in the brain. We reviewed published studies on the subject till December 2007. We precisely localized regions where positive linkage has been found, by the NCBI Map viewer (http://www.ncbi.nlm.nih.gov/mapview/); further, we identified genes located in interesting areas and expressed in the brain, by the Entrez gene, Unigene databases (http://www.ncbi.nlm.nih.gov/entrez/) and Human Protein Reference Database (http://www.hprd.org); these genes could be of interest in future investigations. The review of association studies gave interesting results, as a number of genes seem to be definitively involved in BP, such as SLC6A4, TPH2, DRD4, SLC6A3, DAOA, DTNBP1, NRG1, DISC1 and BDNF. -
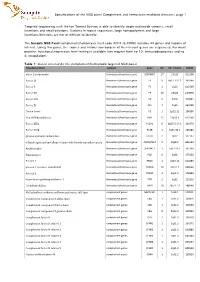
Specifications of the NGS Panel Complement and Hemostasis Mediated Diseases| Page 1
Specifications of the NGS panel Complement and hemostasis mediated diseases| page 1 Targeted sequencing with the Ion Torrent System is able to identify single nucleotide variants, small insertions and small deletions. Variants in repeat sequences, large homopolymers and large insertions/deletions are not or difficult to identify. The Sanquin NGS Panel complement/hemostasis (test code X001 to X006) includes 44 genes and regions of interest. Using this panel, the exones and intron/exon borders of the relevant genes are sequenced. For most proteins, functional/expression level testing is available (see request form no 10: immunodiagnostics and no 4: coagulation). Table 1: Genes covered by the complement/hemostasis targeted NGS panel. Encoded protein Context Gene Chr Chr. Positie OMIM alpha 2-antiplasmin Hemostasis/trombosis gene SERPINF2 17 17p13 613168 Factor IX Hemostasis/trombosis gene F9 X Xq27.1-27.2 300746 Factor V Hemostasis/trombosis gene F5 1 1q23 612309 Factor VII Hemostasis/trombosis gene F7 13 13q34 613878 Factor VIII Hemostasis/trombosis gene F8 X Xq28 300841 Factor XI Hemostasis/trombosis gene F11 4 4q35 264900 Tissue factor Hemostasis/trombosis gene F3 1 1p22-21 134390 Von Willebrand factor Hemostasis/trombosis gene VWF 12 12p13.3 613160 Factor XIIIa Hemostasis/trombosis gene F13A1 6 6p25.3-24.3 134570 Factor XIIIb Hemostasis/trombosis gene F13B 1 1q31-32.1 134580 gamma-glutamyl carboxylase Hemostasis/trombosis gene GGCX 2 2p12 137167 a Disintergrin and metalloproteinase wih thrombospondin repeats Hemostasis/trombosis gene -

Interactive Tutorial 1: SNP Database Resources
NIEHS SNPs Workshop January 30-31, 2005 Interactive Tutorial 1: SNP Database Resources The tutorial is designed to take you through the steps necessary to access SNP data from the primary database resources: 1. Entrez SNP/dbSNP 2. HapMap Genome Browser 3. NIEHS - GeneSNPs and the NIEHS SNPs Websites 4. Other Tools –PolyPhen, ECR, PolyDoms, Transfac Note: Answers to questions from this tutorial are included at the end of this document As a launching point, we will begin our searching at the Entrez cross-database browser. This can be accessed on the NCBI home page (http://www.ncbi.nlm.nih.gov/). For these exercises we will be accessing data for the gene: chemokine-like factor (HUGO name: NOS2A). For a cross-database search: 1. Enter the gene symbol (NOS2A) into the empty box next to the ‘Search All Databases’, type NOS2A into the empty box and click on the GO button, or simply hit the return key on your keyboard. Which NCBI database gives the most number of results What is the database? Hint: Mouse over on the ‘?’ next to the icon and click for a popup explanation of this database. 2. On the left column note the results returned for the ‘SNP’ and ‘Gene’ database. 3. How many results were returned for the ‘SNP’ and ‘Gene’ database? 4. Why did the ‘Gene’ database return more than one result? Entrez Gene 5. From the cross database search, click on the ‘Gene’ database icon. 6. Click on the result that corresponds to the ‘homo sapiens’ NOS2A gene. 7. NOS2A maps to which chromosome? 8. -

A Case of Meningococcal Sepsis and Meningitis with Complement 7
Case Report Infection & http://dx.doi.org/10.3947/ic.2013.45.1.94 Infect Chemother 2013;45(1):94-98 Chemotherapy pISSN 2093-2340 · eISSN 2092-6448 A Case of Meningococcal Sepsis and Meningitis with Complement 7 Deficiency in a Military Trainee Sung Hoon Sim1*, Jung Yeon Heo2*, Eui-Chong Kim3, and Kang-Won Choe2 1Department of Internal Medicine, Seoul National University Hospital, Seoul; 2Department of Internal Medicine, Armed Forces Capital Hospital, Seongnam; 3Department of Laboratory Medicine, Seoul National University Hospital, Seoul, Korea Complement component 7 (C7) deficiency leads to the loss of complement lytic function, and affected patients show in- creased susceptibility to encapsulated organisms infection, especially Neisseria meningitidis. Recently, we have experi- enced a 20-year-old military trainee with meningococcal sepsis and meningitis who was diagnosed as having C7 deficiency based upon the undetectable serum C7 protein on radioimmunoassay. This case emphasizes that although C7 deficiency is rare immune disorder, it is important to be aware of possibility about late complement deficiency among patients who pres- ent with meningococcal disease. Key Words: Neisseria meningitidis, Complement 7 deficiency, Meningitis, Sepsis Introduction of 20 and college freshmen residing in the dormitories are a well-known high risk group. The incidence of meningococcal Humans are the only host for Neisseria meningitidis which disease increases as those high risk age groups from different colonizes the nasopharynx and it is transmitted from human geological areas start communal living [2]. to human by infected respiratory secretions or saliva via air- Apart from these environmental factors, host factors such as borne respiratory droplets. -

The Impact of Complement Genes on the Risk of Late-Onset Alzheimer's
G C A T T A C G G C A T genes Article The Impact of Complement Genes on the Risk of Late-Onset Alzheimer’s Disease Sarah M. Carpanini 1,2,† , Janet C. Harwood 3,† , Emily Baker 1, Megan Torvell 1,2, The GERAD1 Consortium ‡, Rebecca Sims 3 , Julie Williams 1 and B. Paul Morgan 1,2,* 1 UK Dementia Research Institute at Cardiff University, School of Medicine, Cardiff, CF24 4HQ, UK; [email protected] (S.M.C.); [email protected] (E.B.); [email protected] (M.T.); [email protected] (J.W.) 2 Division of Infection and Immunity, School of Medicine, Systems Immunity Research Institute, Cardiff University, Cardiff, CF14 4XN, UK 3 Division of Psychological Medicine and Clinical Neurosciences, School of Medicine, Cardiff University, Cardiff, CF24 4HQ, UK; [email protected] (J.C.H.); [email protected] (R.S.) * Correspondence: [email protected] † These authors contributed equally to this work. ‡ Data used in the preparation of this article were obtained from the Genetic and Environmental Risk for Alzheimer’s disease (GERAD1) Consortium. As such, the investigators within the GERAD1 consortia contributed to the design and implementation of GERAD1 and/or provided data but did not participate in analysis or writing of this report. A full list of GERAD1 investigators and their affiliations is included in Supplementary File S1. Abstract: Late-onset Alzheimer’s disease (LOAD), the most common cause of dementia, and a huge global health challenge, is a neurodegenerative disease of uncertain aetiology. To deliver Citation: Carpanini, S.M.; Harwood, effective diagnostics and therapeutics, understanding the molecular basis of the disease is essential. -

Datasheet: MCA2659F Product Details
Datasheet: MCA2659F Description: MOUSE ANTI HUMAN CD55:FITC Specificity: CD55 Other names: DAF Format: FITC Product Type: Monoclonal Antibody Clone: Bu14 Isotype: IgG1 Quantity: 0.1 mg Product Details Applications This product has been reported to work in the following applications. This information is derived from testing within our laboratories, peer-reviewed publications or personal communications from the originators. Please refer to references indicated for further information. For general protocol recommendations, please visit www.bio- rad-antibodies.com/protocols. Yes No Not Determined Suggested Dilution Flow Cytometry Neat - 1/20 Where this product has not been tested for use in a particular technique this does not necessarily exclude its use in such procedures. Suggested working dilutions are given as a guide only. It is recommended that the user titrates the product for use in their own system using appropriate negative/positive controls. Target Species Human Product Form Purified IgG conjugated to Fluorescein Isothiocyanate Isomer 1 (FITC) - liquid Max Ex/Em Fluorophore Excitation Max (nm) Emission Max (nm) FITC 490 525 Preparation Purified IgG prepared by affinity chromatography on Protein A from tissue culture supernatant Buffer Solution Phosphate buffered saline Preservative 0.09% Sodium Azide (NaN3) Stabilisers 1% Bovine Serum Albumin Approx. Protein IgG concentration 0.1 mg/ml Concentrations Page 1 of 3 External Database Links UniProt: P08174 Related reagents Entrez Gene: 1604 CD55 Related reagents Synonyms CR, DAF RRID AB_2275855 Specificity Mouse anti Human CD55 antibody, clone Bu14 recognizes human CD55, also known as Decay Accelerating Factor (DAF). CD55 is a glycosylphosphatylinositol (GPI) anchored type I surface protein which prevents complement mediated lysis of autologous cells. -

NCBI Web Resources I: Databases and Entrez
NCBI web resources I: databases and Entrez Yanbin Yin Most materials are downloaded from ftp://ftp.ncbi.nih.gov/pub/education/ 1 Homework assignment 1 • Two parts: • 1: Extract the gene IDs reported in table 1 of http://www.ncbi.nlm.nih.gov/pmc/articles/PMC523881/ Using NCBI batch Entrez to download all refseq protein fasta sequences from Arabidopsis thaliana • 2: Read https://academic.oup.com/nar/article/40/D1/D136/2903327 and use your own language to describe what is NCBI taxonomy database and what it can do. • Write a report (in word or ppt) to explain all the operations and include screen shots; save file with your last name in the file name. Due on 9/12 (send by email) 2 References • NCBI mcbios workshop – ftp://ftp.ncbi.nih.gov/pub/education/mcbios2012/ • NCBI web resource tutorials – ftp://ftp.ncbi.nih.gov/pub/education/tutorials/ • NCBI discovery workshops – ftp://ftp.ncbi.nih.gov/pub/education/discovery_workshop s/NLM/2012/Sept2012/ • NCBI Help Manual – http://www.ncbi.nlm.nih.gov/books/NBK3831/ 3 Youtube • http://www.youtube.com/ncbinlm • Go to www.youtube.com • Search “NCBI tutorial general” 4 Topics • Intro. to NCBI • Selected NCBI Databases • The Entrez system • Hands on practice 5 The National Center for Biotechnology Information Bethesda,MD Created in 1988 as a part of the National Library of Medicine at NIH – Establish public databases – Research in computational biology – Develop software tools for sequence analysis – Disseminate biomedical information 6 GenBank history ftp://ftp.ncbi.nih.gov/genbank/ 7 Molecular Data -

A Fatty Liver Study on Mus Musculus
A fatty liver study on Mus musculus Sergio Picart-Armada ∗1 and Alexandre Perera-Lluna †1 1B2SLab at Polytechnic University of Catalonia ∗[email protected] †[email protected] September 17, 2018 Package FELLA 1.13.0 Contents 1 Introduction ..............................2 1.1 Building the database.......................2 1.2 Note on reproducibility ......................3 2 Enrichment analysis .........................4 2.1 Defining the input and running the enrichment ...........4 2.2 Examining the metabolites ....................6 2.3 Examining the genes .......................8 3 Conclusions .............................. 12 4 Reproducibility ............................ 12 References .............................. 14 A fatty liver study on Mus musculus 1 Introduction This vignette shows the utility of the FELLA package, which is based in a statistically normalised diffusion process (Picart-Armada et al. 2017), on non-human organisms. In particular, we will work on a multi-omic Mus musculus study. The original study (Gogiashvili et al. 2017) presents a mouse model of the non-alcoholic fatty liver disease (NAFLD). Metabolites in liver tissue from leptin-deficient ob/ob mice and wild type mice were compared using Nuclear Magnetic Resonance (NMR). Afterwards, quantitative real-time polymerase chain reaction (qRT-PCR) helped identify changes at the gene expression level. Finally, biological mechanisms behind NAFLD were elucidated by leveraging the data from both omics. 1.1 Building the database The first step is to build the FELLA.DATA -

Holird REPORT
CBA HoliRD REPORT: Ellis-Van Creveld Syndrome Marina Esteban Medina María Peña-Chilet Carlos Loucera Joaquín Dopazo Clinical Bioinformatics Area - FPS Sevilla, January 13, 2020 Collaborators: Dr.Víctor Ruiz Pérez ’s group - U760 CIBERER Research group at Instituto de Investigaciones Biomédicas "Alberto Sols" CSIC-UAM, Madrid. CBA Objectives and methodology: The Holistic Rare Disease project (HoliRD) aims to build Diseases Maps for as many Rare Diseases as possible and to model them to systematize research in drug repurposing. In order to achieve this purpose several databases such as ORPHANET, OMIM, HPO, PubMed, KEGG, STRING, as well as the literature is used to collect all the up-to-date knowledge of the diseases under study and defining a Disease Map that contains the functional relationships among the known disease genes, as well as the functional consequences of their activity. Then, a mechanistic model that accounts for the activity of such map is used. The HiPathia algorithm, which has successfully proven to predict cell activities related to cancer hallmarks (Hidalgo et al., Oncotarget 2017; 8:5160-5178; Hidalgo et al., Biol Direct. 2018;13:16) as well as the effect of protein inhibitions on cell survival (Cubuk et al., Cancer Res. 2018; 78:6059-6072) is used to simulate the activity of the disease map. Finally, machine learning algorithms are used to find other proteins, already target of drugs with another indication, which display a potential causal effect on the activity of the previously defined disease map. The drugs that target these proteins are potential candidates for repurposing. Schematic representation of the method used.