Investigation of the Role of Putative Chaperones in Retinal Degeneration
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Review Article Functional Subunits of Eukaryotic Chaperonin CCT/Tric in Protein Folding
SAGE-Hindawi Access to Research Journal of Amino Acids Volume 2011, Article ID 843206, 16 pages doi:10.4061/2011/843206 Review Article Functional Subunits of Eukaryotic Chaperonin CCT/TRiC in Protein Folding M. Anaul Kabir,1 Wasim Uddin,1 Aswathy Narayanan,1 Praveen Kumar Reddy,1 M. Aman Jairajpuri,2 Fred Sherman,3 and Zulfiqar Ahmad4 1 Molecular Genetics Laboratory, School of Biotechnology, National Institute of Technology Calicut, Kerala 673601, India 2 Department of Biosciences, Jamia Millia Islamia, Jamia Nagar, New Delhi 110025, India 3 Department of Biochemistry and Biophysics, University of Rochester Medical Center, NY 14642, USA 4 Department of Biology, Alabama A&M University, Normal, AL 35762, USA Correspondence should be addressed to M. Anaul Kabir, [email protected] Received 15 February 2011; Accepted 5 April 2011 Academic Editor: Shandar Ahmad Copyright © 2011 M. Anaul Kabir et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Molecular chaperones are a class of proteins responsible for proper folding of a large number of polypeptides in both prokaryotic and eukaryotic cells. Newly synthesized polypeptides are prone to nonspecific interactions, and many of them make toxic aggregates in absence of chaperones. The eukaryotic chaperonin CCT is a large, multisubunit, cylindrical structure having two identical rings stacked back to back. Each ring is composed of eight different but similar subunits and each subunit has three distinct domains. CCT assists folding of actin, tubulin, and numerous other cellular proteins in an ATP-dependent manner. -

The Aryl Hydrocarbon Receptor: Structural Analysis and Activation Mechanisms
The Aryl Hydrocarbon Receptor: Structural Analysis and Activation Mechanisms This thesis is submitted in fulfilment of the requirements for the degree of Doctor of Philosophy in the School of Molecular and Biomedical Sciences (Biochemistry), The University of Adelaide, Australia Fiona Whelan, B.Sc. (Hons) 2009 2 Table of Contents THESIS SUMMARY................................................................................. 6 DECLARATION....................................................................................... 7 PUBLICATIONS ARISING FROM THIS THESIS.................................... 8 ACKNOWLEDGEMENTS...................................................................... 10 ABBREVIATIONS ................................................................................. 12 CHAPTER 1: INTRODUCTION ............................................................. 17 1.1 BHLH.PAS PROTEINS ............................................................................................17 1.1.1 General background..................................................................................17 1.1.2 bHLH.PAS Class I Proteins.........................................................................18 1.2 THE ARYL HYDROCARBON RECEPTOR......................................................................19 1.2.1 Domain Structure and Ligand Activation ..............................................19 1.2.2 AhR Expression and Developmental Activity .......................................21 1.2.3 Mouse AhR Knockout Phenotype ...........................................................23 -

Genomic Correlates of Relationship QTL Involved in Fore- Versus Hind Limb Divergence in Mice
Loyola University Chicago Loyola eCommons Biology: Faculty Publications and Other Works Faculty Publications 2013 Genomic Correlates of Relationship QTL Involved in Fore- Versus Hind Limb Divergence in Mice Mihaela Palicev Gunter P. Wagner James P. Noonan Benedikt Hallgrimsson James M. Cheverud Loyola University Chicago, [email protected] Follow this and additional works at: https://ecommons.luc.edu/biology_facpubs Part of the Biology Commons Recommended Citation Palicev, M, GP Wagner, JP Noonan, B Hallgrimsson, and JM Cheverud. "Genomic Correlates of Relationship QTL Involved in Fore- Versus Hind Limb Divergence in Mice." Genome Biology and Evolution 5(10), 2013. This Article is brought to you for free and open access by the Faculty Publications at Loyola eCommons. It has been accepted for inclusion in Biology: Faculty Publications and Other Works by an authorized administrator of Loyola eCommons. For more information, please contact [email protected]. This work is licensed under a Creative Commons Attribution-Noncommercial-No Derivative Works 3.0 License. © Palicev et al., 2013. GBE Genomic Correlates of Relationship QTL Involved in Fore- versus Hind Limb Divergence in Mice Mihaela Pavlicev1,2,*, Gu¨ nter P. Wagner3, James P. Noonan4, Benedikt Hallgrı´msson5,and James M. Cheverud6 1Konrad Lorenz Institute for Evolution and Cognition Research, Altenberg, Austria 2Department of Pediatrics, Cincinnati Children‘s Hospital Medical Center, Cincinnati, Ohio 3Yale Systems Biology Institute and Department of Ecology and Evolutionary Biology, Yale University 4Department of Genetics, Yale University School of Medicine 5Department of Cell Biology and Anatomy, The McCaig Institute for Bone and Joint Health and the Alberta Children’s Hospital Research Institute for Child and Maternal Health, University of Calgary, Calgary, Canada 6Department of Anatomy and Neurobiology, Washington University *Corresponding author: E-mail: [email protected]. -

Germline Mutations Affecting Gα11 in Hypoparathyroidism
T h e new england journal o f medicine of a meta-analysis of studies of the effect of re- terone levels were increased. Although long- duced dietary salt on the incidence of cardiovas- term, modest reductions in salt intake result in cular events and death.1 The authors of the Co- small, physiologic increases in plasma renin ac- chrane report wrote that there was “no strong tivity,3 the preponderance of evidence suggests evidence of benefit.” In a summary statement, we that a reduced salt intake is associated with a wrote that this particular Cochrane analysis con- decreased risk of cardiovascular events and cluded that reducing dietary salt intake did not death. Furthermore, it is worth remembering decrease the risk of death or cardiovascular dis- that diuretics remain one of the most effective ease. Stigler et al. suggest that “indeterminate antihypertensive therapies, and their beneficial results,” rather than no significant effect, would effect on cardiovascular disease is well docu- be a more appropriate interpretation of the analy- mented.4 Nevertheless, as we suggested, in sis. Both interpretations may be correct. Although terms of safety, the lower limit of salt consump- it may not be possible to reject the null hypothe- tion has not been clearly defined. sis with certainty (i.e., no effect of reduced salt), Theodore A. Kotchen, M.D. the analysis should have been powered to detect Allen W. Cowley, Jr., Ph.D. a clinically meaningful difference, and the con- Medical College of Wisconsin clusion of no effect provides more guidance than Milwaukee, WI “indeterminate results” to clinical decision mak- [email protected] ers. -

S41598-018-28214-2.Pdf
www.nature.com/scientificreports OPEN Dissecting Distinct Roles of NEDDylation E1 Ligase Heterodimer APPBP1 and UBA3 Received: 7 November 2017 Accepted: 25 May 2018 Reveals Potential Evolution Process Published: xx xx xxxx for Activation of Ubiquitin-related Pathways Harbani Kaur Malik-Chaudhry1, Zied Gaieb1, Amanda Saavedra1, Michael Reyes1, Raphael Kung1, Frank Le1, Dimitrios Morikis1,2 & Jiayu Liao1,2 Despite the similar enzyme cascade in the Ubiquitin and Ubiquitin-like peptide(Ubl) conjugation, the involvement of single or heterodimer E1 activating enzyme has been a mystery. Here, by using a quantitative Förster Resonance Energy Transfer (FRET) technology, aided with Analysis of Electrostatic Similarities Of Proteins (AESOP) computational framework, we elucidate in detail the functional properties of each subunit of the E1 heterodimer activating-enzyme for NEDD8, UBA3 and APPBP1. In contrast to SUMO activation, which requires both subunits of its E1 heterodimer AOS1-Uba2 for its activation, NEDD8 activation requires only one of two E1 subunits, UBA3. The other subunit, APPBP1, only contributes by accelerating the activation reaction rate. This discovery implies that APPBP1 functions mainly as a scafold protein to enhance molecular interactions and facilitate catalytic reaction. These fndings for the frst time reveal critical new mechanisms and a potential evolutionary pathway for Ubl activations. Furthermore, this quantitative FRET approach can be used for other general biochemical pathway analysis in a dynamic mode. Ubiquitin and Ubls are peptides that are conjugated to various target proteins to either lead the targeted protein to degradation or changes of activities in vivo, and their dysregulations ofen leads to various diseases, such as can- cers or neurodegenerative diseases1–3. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Receptor-Arrestin Interactions: the GPCR Perspective
biomolecules Review Receptor-Arrestin Interactions: The GPCR Perspective Mohammad Seyedabadi 1,2 , Mehdi Gharghabi 3, Eugenia V. Gurevich 4 and Vsevolod V. Gurevich 4,* 1 Department of Toxicology & Pharmacology, Faculty of Pharmacy, Mazandaran University of Medical Sciences, Sari 48471-93698, Iran; [email protected] 2 Pharmaceutical Sciences Research Center, Faculty of Pharmacy, Mazandaran University of Medical Sciences, Sari 48167-75952, Iran 3 Department of Cancer Biology and Genetics, The Ohio State University Wexner Medical Center, Columbus, OH 43210, USA; [email protected] 4 Department of Pharmacology, Vanderbilt University, Nashville, TN 37232, USA; [email protected] * Correspondence: [email protected]; Tel.: +1-615-322-7070; Fax: +1-615-343-6532 Abstract: Arrestins are a small family of four proteins in most vertebrates that bind hundreds of different G protein-coupled receptors (GPCRs). Arrestin binding to a GPCR has at least three functions: precluding further receptor coupling to G proteins, facilitating receptor internalization, and initiating distinct arrestin-mediated signaling. The molecular mechanism of arrestin–GPCR interactions has been extensively studied and discussed from the “arrestin perspective”, focusing on the roles of arrestin elements in receptor binding. Here, we discuss this phenomenon from the “receptor perspective”, focusing on the receptor elements involved in arrestin binding and empha- sizing existing gaps in our knowledge that need to be filled. It is vitally important to understand the role of receptor elements in arrestin activation and how the interaction of each of these elements with arrestin contributes to the latter’s transition to the high-affinity binding state. A more precise knowledge of the molecular mechanisms of arrestin activation is needed to enable the construction of arrestin mutants with desired functional characteristics. -

Ubiquitylome Profiling of Parkin-Null Brain Reveals Dysregulation Of
Neurobiology of Disease 127 (2019) 114–130 Contents lists available at ScienceDirect Neurobiology of Disease journal homepage: www.elsevier.com/locate/ynbdi Ubiquitylome profiling of Parkin-null brain reveals dysregulation of calcium T homeostasis factors ATP1A2, Hippocalcin and GNA11, reflected by altered firing of noradrenergic neurons Key J.a,1, Mueller A.K.b,1, Gispert S.a, Matschke L.b, Wittig I.c, Corti O.d,e,f,g, Münch C.h, ⁎ ⁎ Decher N.b, , Auburger G.a, a Exp. Neurology, Goethe University Medical School, 60590 Frankfurt am Main, Germany b Institute for Physiology and Pathophysiology, Vegetative Physiology and Marburg Center for Mind, Brain and Behavior - MCMBB; Clinic for Neurology, Philipps-University Marburg, 35037 Marburg, Germany c Functional Proteomics, SFB 815 Core Unit, Goethe University Medical School, 60590 Frankfurt am Main, Germany d Institut du Cerveau et de la Moelle épinière, ICM, Paris, F-75013, France e Inserm, U1127, Paris, F-75013, France f CNRS, UMR 7225, Paris, F-75013, France g Sorbonne Universités, Paris, F-75013, France h Institute of Biochemistry II, Goethe University Medical School, 60590 Frankfurt am Main, Germany ARTICLE INFO ABSTRACT Keywords: Parkinson's disease (PD) is the second most frequent neurodegenerative disorder in the old population. Among Parkinson's disease its monogenic variants, a frequent cause is a mutation in the Parkin gene (Prkn). Deficient function of Parkin Mitochondria triggers ubiquitous mitochondrial dysfunction and inflammation in the brain, but it remains unclear howse- Parkin lective neural circuits become vulnerable and finally undergo atrophy. Ubiquitin We attempted to go beyond previous work, mostly done in peripheral tumor cells, which identified protein Calcium targets of Parkin activity, an ubiquitin E3 ligase. -
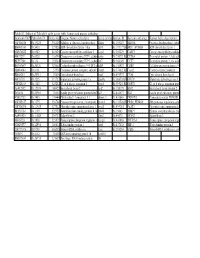
Table S3: Subset of Zebrafish Early Genes with Human And
Table S3: Subset of Zebrafish early genes with human and mouse orthologs Genbank ID(ZFZebrafish ID Entrez GenUnigene Name (zebrafish) Gene symbo Human ID Humann ortholog Human Gene description AW116838 Dr.19225 336425 Aldolase a, fructose-bisphosphate aldoa Hs.155247 ALDOA Fructose-bisphosphate aldola BM005100 Dr.5438 327026 ADP-ribosylation factor 1 like arf1l Hs.119177||HsARF1_HUMAN ADP-ribosylation factor 1 AW076882 Dr.6582 403025 Cancer susceptibility candidate 3 casc3 Hs.350229 CASC3 Cancer susceptibility candidat AI437239 Dr.6928 116994 Chaperonin containing TCP1, subun cct6a Hs.73072||Hs.CCT6A T-complex protein 1, zeta sub BE557308 Dr.134 192324 Chaperonin containing TCP1, subun cct7 Hs.368149 CCT7 T-complex protein 1, eta subu BG303647 Dr.26326 321602 Cyclin-dependent kinase 9 (CDC2-recdk9 Hs.150423 CDK9 Cell division protein kinase 9 AB040044 Dr.8161 57970 Coatomer protein complex, subunit zcopz1 Hs.37482||Hs.Copz2 Coatomer zeta-2 subunit BI888253 Dr.20911 30436 Eyes absent homolog 1 eya1 Hs.491997 EYA4 Eyes absent homolog 4 AI878758 Dr.3225 317737 Glutamate dehydrogenase 1a glud1a Hs.368538||HsGLUD1 Glutamate dehydrogenase 1, AW128619 Dr.1388 325284 G1 to S phase transition 1 gspt1 Hs.59523||Hs.GSPT1 G1 to S phase transition prote AF412832 Dr.12595 140427 Heat shock factor 2 hsf2 Hs.158195 HSF2 Heat shock factor protein 2 D38454 Dr.20916 30151 Insulin gene enhancer protein Islet3 isl3 Hs.444677 ISL2 Insulin gene enhancer protein AY052752 Dr.7485 170444 Pbx/knotted 1 homeobox 1.1 pknox1.1 Hs.431043 PKNOX1 Homeobox protein PKNOX1 -

Predicting Coupling Probabilities of G-Protein Coupled Receptors Gurdeep Singh1,2,†, Asuka Inoue3,*,†, J
Published online 30 May 2019 Nucleic Acids Research, 2019, Vol. 47, Web Server issue W395–W401 doi: 10.1093/nar/gkz392 PRECOG: PREdicting COupling probabilities of G-protein coupled receptors Gurdeep Singh1,2,†, Asuka Inoue3,*,†, J. Silvio Gutkind4, Robert B. Russell1,2,* and Francesco Raimondi1,2,* 1CellNetworks, Bioquant, Heidelberg University, Im Neuenheimer Feld 267, 69120 Heidelberg, Germany, 2Biochemie Zentrum Heidelberg (BZH), Heidelberg University, Im Neuenheimer Feld 328, 69120 Heidelberg, Germany, 3Graduate School of Pharmaceutical Sciences, Tohoku University, Sendai, Miyagi 980-8578, Japan and 4Department of Pharmacology and Moores Cancer Center, University of California, San Diego, La Jolla, CA 92093, USA Received February 10, 2019; Revised April 13, 2019; Editorial Decision April 24, 2019; Accepted May 01, 2019 ABSTRACT great use in tinkering with signalling pathways in living sys- tems (5). G-protein coupled receptors (GPCRs) control multi- Ligand binding to GPCRs induces conformational ple physiological states by transducing a multitude changes that lead to binding and activation of G-proteins of extracellular stimuli into the cell via coupling to situated on the inner cell membrane. Most of mammalian intra-cellular heterotrimeric G-proteins. Deciphering GPCRs couple with more than one G-protein giving each which G-proteins couple to each of the hundreds receptor a distinct coupling profile (6) and thus specific of GPCRs present in a typical eukaryotic organism downstream cellular responses. Determining these coupling is therefore critical to understand signalling. Here, profiles is critical to understand GPCR biology and phar- we present PRECOG (precog.russelllab.org): a web- macology. Despite decades of research and hundreds of ob- server for predicting GPCR coupling, which allows served interactions, coupling information is still missing for users to: (i) predict coupling probabilities for GPCRs many receptors and sequence determinants of coupling- specificity are still largely unknown. -

When 7 Transmembrane Receptors Are Not G Protein–Coupled Receptors
When 7 transmembrane receptors are not G protein–coupled receptors Keshava Rajagopal, … , Robert J. Lefkowitz, Howard A. Rockman J Clin Invest. 2005;115(11):2971-2974. https://doi.org/10.1172/JCI26950. Commentary Classically, 7 transmembrane receptors transduce extracellular signals by coupling to heterotrimeric G proteins, although recent in vitro studies have clearly demonstrated that they can also signal via G protein–independent mechanisms. However, the physiologic consequences of this unconventional signaling, particularly in vivo, have not been explored. In this issue of the JCI, Zhai et al. demonstrate in vivo effects of G protein–independent signaling by the angiotensin II type 1 receptor (AT1R). In studies of the mouse heart, they compare the physiologic and biochemical consequences of transgenic cardiac-specific overexpression of a mutant AT1R incapable of G protein coupling with those of a wild-type receptor. Their results not only provide the first glimpse of the physiologic effects of this newly appreciated mode of signaling but also provide important and previously unappreciated clues as to the underlying molecular mechanisms. Find the latest version: https://jci.me/26950/pdf commentaries gene, spastin, regulates microtubule stability to 16. Wittmann, C.W., et al. 2001. Tauopathy in Dro- 21. Chan, Y.B., et al. 2003. Neuromuscular defects in modulate synaptic structure and function. Curr. sophila: neurodegeneration without neurofibril- a Drosophila survival motor neuron gene mutant. Biol. 14:1135–1147. lary tangles. Science. 293:711–714. Hum. Mol. Genet. 12:1367–1376. 12. Sherwood, N.T., Sun, Q., Xue, M., Zhang, B., and 17. Shapiro, W.R., and Young, D.F. -

Supplemental Information
Supplemental information Dissection of the genomic structure of the miR-183/96/182 gene. Previously, we showed that the miR-183/96/182 cluster is an intergenic miRNA cluster, located in a ~60-kb interval between the genes encoding nuclear respiratory factor-1 (Nrf1) and ubiquitin-conjugating enzyme E2H (Ube2h) on mouse chr6qA3.3 (1). To start to uncover the genomic structure of the miR- 183/96/182 gene, we first studied genomic features around miR-183/96/182 in the UCSC genome browser (http://genome.UCSC.edu/), and identified two CpG islands 3.4-6.5 kb 5’ of pre-miR-183, the most 5’ miRNA of the cluster (Fig. 1A; Fig. S1 and Seq. S1). A cDNA clone, AK044220, located at 3.2-4.6 kb 5’ to pre-miR-183, encompasses the second CpG island (Fig. 1A; Fig. S1). We hypothesized that this cDNA clone was derived from 5’ exon(s) of the primary transcript of the miR-183/96/182 gene, as CpG islands are often associated with promoters (2). Supporting this hypothesis, multiple expressed sequences detected by gene-trap clones, including clone D016D06 (3, 4), were co-localized with the cDNA clone AK044220 (Fig. 1A; Fig. S1). Clone D016D06, deposited by the German GeneTrap Consortium (GGTC) (http://tikus.gsf.de) (3, 4), was derived from insertion of a retroviral construct, rFlpROSAβgeo in 129S2 ES cells (Fig. 1A and C). The rFlpROSAβgeo construct carries a promoterless reporter gene, the β−geo cassette - an in-frame fusion of the β-galactosidase and neomycin resistance (Neor) gene (5), with a splicing acceptor (SA) immediately upstream, and a polyA signal downstream of the β−geo cassette (Fig.