Molecular Mechanics - Force Field Method
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Key Concepts for Future QIS Learners Workshop Output Published Online May 13, 2020
Key Concepts for Future QIS Learners Workshop output published online May 13, 2020 Background and Overview On behalf of the Interagency Working Group on Workforce, Industry and Infrastructure, under the NSTC Subcommittee on Quantum Information Science (QIS), the National Science Foundation invited 25 researchers and educators to come together to deliberate on defining a core set of key concepts for future QIS learners that could provide a starting point for further curricular and educator development activities. The deliberative group included university and industry researchers, secondary school and college educators, and representatives from educational and professional organizations. The workshop participants focused on identifying concepts that could, with additional supporting resources, help prepare secondary school students to engage with QIS and provide possible pathways for broader public engagement. This workshop report identifies a set of nine Key Concepts. Each Concept is introduced with a concise overall statement, followed by a few important fundamentals. Connections to current and future technologies are included, providing relevance and context. The first Key Concept defines the field as a whole. Concepts 2-6 introduce ideas that are necessary for building an understanding of quantum information science and its applications. Concepts 7-9 provide short explanations of critical areas of study within QIS: quantum computing, quantum communication and quantum sensing. The Key Concepts are not intended to be an introductory guide to quantum information science, but rather provide a framework for future expansion and adaptation for students at different levels in computer science, mathematics, physics, and chemistry courses. As such, it is expected that educators and other community stakeholders may not yet have a working knowledge of content covered in the Key Concepts. -

GROMACS: Fast, Flexible, and Free
GROMACS: Fast, Flexible, and Free DAVID VAN DER SPOEL,1 ERIK LINDAHL,2 BERK HESS,3 GERRIT GROENHOF,4 ALAN E. MARK,4 HERMAN J. C. BERENDSEN4 1Department of Cell and Molecular Biology, Uppsala University, Husargatan 3, Box 596, S-75124 Uppsala, Sweden 2Stockholm Bioinformatics Center, SCFAB, Stockholm University, SE-10691 Stockholm, Sweden 3Max-Planck Institut fu¨r Polymerforschung, Ackermannweg 10, D-55128 Mainz, Germany 4Groningen Biomolecular Sciences and Biotechnology Institute, University of Groningen, Nijenborgh 4, NL-9747 AG Groningen, The Netherlands Received 12 February 2005; Accepted 18 March 2005 DOI 10.1002/jcc.20291 Published online in Wiley InterScience (www.interscience.wiley.com). Abstract: This article describes the software suite GROMACS (Groningen MAchine for Chemical Simulation) that was developed at the University of Groningen, The Netherlands, in the early 1990s. The software, written in ANSI C, originates from a parallel hardware project, and is well suited for parallelization on processor clusters. By careful optimization of neighbor searching and of inner loop performance, GROMACS is a very fast program for molecular dynamics simulation. It does not have a force field of its own, but is compatible with GROMOS, OPLS, AMBER, and ENCAD force fields. In addition, it can handle polarizable shell models and flexible constraints. The program is versatile, as force routines can be added by the user, tabulated functions can be specified, and analyses can be easily customized. Nonequilibrium dynamics and free energy determinations are incorporated. Interfaces with popular quantum-chemical packages (MOPAC, GAMES-UK, GAUSSIAN) are provided to perform mixed MM/QM simula- tions. The package includes about 100 utility and analysis programs. -
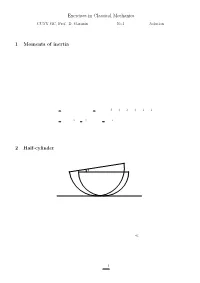
Exercises in Classical Mechanics 1 Moments of Inertia 2 Half-Cylinder
Exercises in Classical Mechanics CUNY GC, Prof. D. Garanin No.1 Solution ||||||||||||||||||||||||||||||||||||||||| 1 Moments of inertia (5 points) Calculate tensors of inertia with respect to the principal axes of the following bodies: a) Hollow sphere of mass M and radius R: b) Cone of the height h and radius of the base R; both with respect to the apex and to the center of mass. c) Body of a box shape with sides a; b; and c: Solution: a) By symmetry for the sphere I®¯ = I±®¯: (1) One can ¯nd I easily noticing that for the hollow sphere is 1 1 X ³ ´ I = (I + I + I ) = m y2 + z2 + z2 + x2 + x2 + y2 3 xx yy zz 3 i i i i i i i i 2 X 2 X 2 = m r2 = R2 m = MR2: (2) 3 i i 3 i 3 i i b) and c): Standard solutions 2 Half-cylinder ϕ (10 points) Consider a half-cylinder of mass M and radius R on a horizontal plane. a) Find the position of its center of mass (CM) and the moment of inertia with respect to CM. b) Write down the Lagrange function in terms of the angle ' (see Fig.) c) Find the frequency of cylinder's oscillations in the linear regime, ' ¿ 1. Solution: (a) First we ¯nd the distance a between the CM and the geometrical center of the cylinder. With σ being the density for the cross-sectional surface, so that ¼R2 M = σ ; (3) 2 1 one obtains Z Z 2 2σ R p σ R q a = dx x R2 ¡ x2 = dy R2 ¡ y M 0 M 0 ¯ 2 ³ ´3=2¯R σ 2 2 ¯ σ 2 3 4 = ¡ R ¡ y ¯ = R = R: (4) M 3 0 M 3 3¼ The moment of inertia of the half-cylinder with respect to the geometrical center is the same as that of the cylinder, 1 I0 = MR2: (5) 2 The moment of inertia with respect to the CM I can be found from the relation I0 = I + Ma2 (6) that yields " µ ¶ # " # 1 4 2 1 32 I = I0 ¡ Ma2 = MR2 ¡ = MR2 1 ¡ ' 0:3199MR2: (7) 2 3¼ 2 (3¼)2 (b) The Lagrange function L is given by L('; '_ ) = T ('; '_ ) ¡ U('): (8) The potential energy U of the half-cylinder is due to the elevation of its CM resulting from the deviation of ' from zero. -

Development of an Advanced Force Field for Water Using Variational Energy Decomposition Analysis Arxiv:1905.07816V3 [Physics.Ch
Development of an Advanced Force Field for Water using Variational Energy Decomposition Analysis Akshaya K. Das,y Lars Urban,y Itai Leven,y Matthias Loipersberger,y Abdulrahman Aldossary,y Martin Head-Gordon,y and Teresa Head-Gordon∗,z y Pitzer Center for Theoretical Chemistry, Department of Chemistry, University of California, Berkeley CA 94720 z Pitzer Center for Theoretical Chemistry, Departments of Chemistry, Bioengineering, Chemical and Biomolecular Engineering, University of California, Berkeley CA 94720 E-mail: [email protected] Abstract Given the piecewise approach to modeling intermolecular interactions for force fields, they can be difficult to parameterize since they are fit to data like total en- ergies that only indirectly connect to their separable functional forms. Furthermore, by neglecting certain types of molecular interactions such as charge penetration and charge transfer, most classical force fields must rely on, but do not always demon- arXiv:1905.07816v3 [physics.chem-ph] 27 Jul 2019 strate, how cancellation of errors occurs among the remaining molecular interactions accounted for such as exchange repulsion, electrostatics, and polarization. In this work we present the first generation of the (many-body) MB-UCB force field that explicitly accounts for the decomposed molecular interactions commensurate with a variational energy decomposition analysis, including charge transfer, with force field design choices 1 that reduce the computational expense of the MB-UCB potential while remaining accu- rate. We optimize parameters using only single water molecule and water cluster data up through pentamers, with no fitting to condensed phase data, and we demonstrate that high accuracy is maintained when the force field is subsequently validated against conformational energies of larger water cluster data sets, radial distribution functions of the liquid phase, and the temperature dependence of thermodynamic and transport water properties. -

FORCE FIELDS for PROTEIN SIMULATIONS by JAY W. PONDER
FORCE FIELDS FOR PROTEIN SIMULATIONS By JAY W. PONDER* AND DAVIDA. CASEt *Department of Biochemistry and Molecular Biophysics, Washington University School of Medicine, 51. Louis, Missouri 63110, and tDepartment of Molecular Biology, The Scripps Research Institute, La Jolla, California 92037 I. Introduction. ...... .... ... .. ... .... .. .. ........ .. .... .... ........ ........ ..... .... 27 II. Protein Force Fields, 1980 to the Present.............................................. 30 A. The Am.ber Force Fields.............................................................. 30 B. The CHARMM Force Fields ..., ......... 35 C. The OPLS Force Fields............................................................... 38 D. Other Protein Force Fields ....... 39 E. Comparisons Am.ong Protein Force Fields ,... 41 III. Beyond Fixed Atomic Point-Charge Electrostatics.................................... 45 A. Limitations of Fixed Atomic Point-Charges ........ 46 B. Flexible Models for Static Charge Distributions.................................. 48 C. Including Environmental Effects via Polarization................................ 50 D. Consistent Treatment of Electrostatics............................................. 52 E. Current Status of Polarizable Force Fields........................................ 57 IV. Modeling the Solvent Environment .... 62 A. Explicit Water Models ....... 62 B. Continuum Solvent Models.......................................................... 64 C. Molecular Dynamics Simulations with the Generalized Born Model........ -

Force Fields for MD Simulations
Force Fields for MD simulations • Topology/parameter files • Where do the numbers an MD code uses come from? • How to make topology files for ligands, cofactors, special amino acids, … • How to obtain/develop missing parameters. • QM and QM/MM force fields/potential energy descriptions used for molecular simulations. The Potential Energy Function Ubond = oscillations about the equilibrium bond length Uangle = oscillations of 3 atoms about an equilibrium bond angle Udihedral = torsional rotation of 4 atoms about a central bond Unonbond = non-bonded energy terms (electrostatics and Lenard-Jones) Energy Terms Described in the CHARMm Force Field Bond Angle Dihedral Improper Classical Molecular Dynamics r(t +!t) = r(t) + v(t)!t v(t +!t) = v(t) + a(t)!t a(t) = F(t)/ m d F = ! U (r) dr Classical Molecular Dynamics 12 6 &, R ) , R ) # U (r) = . $* min,ij ' - 2* min,ij ' ! 1 qiq j ij * ' * ' U (r) = $ rij rij ! %+ ( + ( " 4!"0 rij Coulomb interaction van der Waals interaction Classical Molecular Dynamics Classical Molecular Dynamics Bond definitions, atom types, atom names, parameters, …. What is a Force Field? In molecular dynamics a molecule is described as a series of charged points (atoms) linked by springs (bonds). To describe the time evolution of bond lengths, bond angles and torsions, also the non-bonding van der Waals and elecrostatic interactions between atoms, one uses a force field. The force field is a collection of equations and associated constants designed to reproduce molecular geometry and selected properties of tested structures. Energy Functions Ubond = oscillations about the equilibrium bond length Uangle = oscillations of 3 atoms about an equilibrium bond angle Udihedral = torsional rotation of 4 atoms about a central bond Unonbond = non-bonded energy terms (electrostatics and Lenard-Jones) Parameter optimization of the CHARMM Force Field Based on the protocol established by Alexander D. -

Quantum Mechanics
Quantum Mechanics Richard Fitzpatrick Professor of Physics The University of Texas at Austin Contents 1 Introduction 5 1.1 Intendedaudience................................ 5 1.2 MajorSources .................................. 5 1.3 AimofCourse .................................. 6 1.4 OutlineofCourse ................................ 6 2 Probability Theory 7 2.1 Introduction ................................... 7 2.2 WhatisProbability?.............................. 7 2.3 CombiningProbabilities. ... 7 2.4 Mean,Variance,andStandardDeviation . ..... 9 2.5 ContinuousProbabilityDistributions. ........ 11 3 Wave-Particle Duality 13 3.1 Introduction ................................... 13 3.2 Wavefunctions.................................. 13 3.3 PlaneWaves ................................... 14 3.4 RepresentationofWavesviaComplexFunctions . ....... 15 3.5 ClassicalLightWaves ............................. 18 3.6 PhotoelectricEffect ............................. 19 3.7 QuantumTheoryofLight. .. .. .. .. .. .. .. .. .. .. .. .. .. 21 3.8 ClassicalInterferenceofLightWaves . ...... 21 3.9 QuantumInterferenceofLight . 22 3.10 ClassicalParticles . .. .. .. .. .. .. .. .. .. .. .. .. .. .. 25 3.11 QuantumParticles............................... 25 3.12 WavePackets .................................. 26 2 QUANTUM MECHANICS 3.13 EvolutionofWavePackets . 29 3.14 Heisenberg’sUncertaintyPrinciple . ........ 32 3.15 Schr¨odinger’sEquation . 35 3.16 CollapseoftheWaveFunction . 36 4 Fundamentals of Quantum Mechanics 39 4.1 Introduction .................................. -

FORCE FIELDS and CRYSTAL STRUCTURE PREDICTION Contents
FORCE FIELDS AND CRYSTAL STRUCTURE PREDICTION Bouke P. van Eijck ([email protected]) Utrecht University (Retired) Department of Crystal and Structural Chemistry Padualaan 8, 3584 CH Utrecht, The Netherlands Originally written in 2003 Update blind tests 2017 Contents 1 Introduction 2 2 Lattice Energy 2 2.1 Polarcrystals .............................. 4 2.2 ConvergenceAcceleration . 5 2.3 EnergyMinimization .......................... 6 3 Temperature effects 8 3.1 LatticeVibrations............................ 8 4 Prediction of Crystal Structures 9 4.1 Stage1:generationofpossiblestructures . .... 9 4.2 Stage2:selectionoftherightstructure(s) . ..... 11 4.3 Blindtests................................ 14 4.4 Beyondempiricalforcefields. 15 4.5 Conclusions............................... 17 4.6 Update2017............................... 17 1 1 Introduction Everybody who looks at a crystal structure marvels how Nature finds a way to pack complex molecules into space-filling patterns. The question arises: can we understand such packings without doing experiments? This is a great challenge to theoretical chemistry. Most work in this direction uses the concept of a force field. This is just the po- tential energy of a collection of atoms as a function of their coordinates. In principle, this energy can be calculated by quantumchemical methods for a free molecule; even for an entire crystal computations are beginning to be feasible. But for nearly all work a parameterized functional form for the energy is necessary. An ab initio force field is derived from the abovementioned calculations on small model systems, which can hopefully be generalized to other related substances. This is a relatively new devel- opment, and most force fields are empirical: they have been developed to reproduce observed properties as well as possible. There exists a number of more or less time- honored force fields: MM3, CHARMM, AMBER, GROMOS, OPLS, DREIDING.. -

Newtonian Mechanics Is Most Straightforward in Its Formulation and Is Based on Newton’S Second Law
CLASSICAL MECHANICS D. A. Garanin September 30, 2015 1 Introduction Mechanics is part of physics studying motion of material bodies or conditions of their equilibrium. The latter is the subject of statics that is important in engineering. General properties of motion of bodies regardless of the source of motion (in particular, the role of constraints) belong to kinematics. Finally, motion caused by forces or interactions is the subject of dynamics, the biggest and most important part of mechanics. Concerning systems studied, mechanics can be divided into mechanics of material points, mechanics of rigid bodies, mechanics of elastic bodies, and mechanics of fluids: hydro- and aerodynamics. At the core of each of these areas of mechanics is the equation of motion, Newton's second law. Mechanics of material points is described by ordinary differential equations (ODE). One can distinguish between mechanics of one or few bodies and mechanics of many-body systems. Mechanics of rigid bodies is also described by ordinary differential equations, including positions and velocities of their centers and the angles defining their orientation. Mechanics of elastic bodies and fluids (that is, mechanics of continuum) is more compli- cated and described by partial differential equation. In many cases mechanics of continuum is coupled to thermodynamics, especially in aerodynamics. The subject of this course are systems described by ODE, including particles and rigid bodies. There are two limitations on classical mechanics. First, speeds of the objects should be much smaller than the speed of light, v c, otherwise it becomes relativistic mechanics. Second, the bodies should have a sufficiently large mass and/or kinetic energy. -

Rotation: Moment of Inertia and Torque
Rotation: Moment of Inertia and Torque Every time we push a door open or tighten a bolt using a wrench, we apply a force that results in a rotational motion about a fixed axis. Through experience we learn that where the force is applied and how the force is applied is just as important as how much force is applied when we want to make something rotate. This tutorial discusses the dynamics of an object rotating about a fixed axis and introduces the concepts of torque and moment of inertia. These concepts allows us to get a better understanding of why pushing a door towards its hinges is not very a very effective way to make it open, why using a longer wrench makes it easier to loosen a tight bolt, etc. This module begins by looking at the kinetic energy of rotation and by defining a quantity known as the moment of inertia which is the rotational analog of mass. Then it proceeds to discuss the quantity called torque which is the rotational analog of force and is the physical quantity that is required to changed an object's state of rotational motion. Moment of Inertia Kinetic Energy of Rotation Consider a rigid object rotating about a fixed axis at a certain angular velocity. Since every particle in the object is moving, every particle has kinetic energy. To find the total kinetic energy related to the rotation of the body, the sum of the kinetic energy of every particle due to the rotational motion is taken. The total kinetic energy can be expressed as .. -

Energy Functions and Their Relationship to Molecular Conformation
Molecular dynamics simulation CS/CME/BioE/Biophys/BMI 279 Oct. 5, 2021 Ron Dror 1 Outline • Molecular dynamics (MD): The basic idea • Equations of motion • Key properties of MD simulations • Sample applications • Limitations of MD simulations • Software packages and force fields • Accelerating MD simulations • Monte Carlo simulation 2 Molecular dynamics: The basic idea 3 The idea • Mimic what atoms do in real life, assuming a given potential energy function – The energy function allows us to calculate the force experienced by any atom given the positions of the other atoms – Newton’s laws tell us how those forces will affect the motions of the atoms ) U Energy( Position Position 4 Basic algorithm • Divide time into discrete time steps, no more than a few femtoseconds (10–15 s) each • At each time step: – Compute the forces acting on each atom, using a molecular mechanics force field – Move the atoms a little bit: update position and velocity of each atom using Newton’s laws of motion 5 Molecular dynamics movie Equations of motion 7 Specifying atom positions • For a system with N x1 atoms, we can specify y1 the position of all z1 atoms by a single x2 vector x of length 3N y2 x = – This vector contains z2 the x, y, and z ⋮ coordinates of every xN atom yN zN Specifying forces ∂U • A single vector F specifies F1,x ∂x1 the force acting on every F ∂U atom in the system 1,y ∂y1 • For a system with N F ∂U atoms, F is a vector of 1,z ∂z1 length 3N ∂U F2,x – This vector lists the force ∂x2 ∂U on each atom in the x-, y-, F2,y and z- directions F -

Molecular Docking : Current Advances and Challenges
ARTÍCULO DE REVISIÓN © 2018 Universidad Nacional Autónoma de México, Facultad de Estudios Superiores Zaragoza. This is an Open Access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/). TIP Revista Especializada en Ciencias Químico-Biológicas, 21(Supl. 1): 65-87, 2018. DOI: 10.22201/fesz.23958723e.2018.0.143 MOLECULAR DOCKING: CURRENT ADVANCES AND CHALLENGES Fernando D. Prieto-Martínez,1a Marcelino Arciniega2 and José L. Medina-Franco1b 1Facultad de Química, Departamento de Farmacia, Universidad Nacional Autónoma de México, Avenida Universidad # 3000, Delegación Coyoacán, Ciudad de México 04510, México 2Instituto de Fisiología Celular, Departamento de Bioquímica y Biología Estructural, Universidad Nacional Autónoma de México E-mails: [email protected], [email protected],[email protected] ABSTRACT Automated molecular docking aims at predicting the possible interactions between two molecules. This method has proven useful in medicinal chemistry and drug discovery providing atomistic insights into molecular recognition. Over the last 20 years methods for molecular docking have been improved, yielding accurate results on pose prediction. Nonetheless, several aspects of molecular docking need revision due to changes in the paradigm of drug discovery. In the present article, we review the principles, techniques, and algorithms for docking with emphasis on protein-ligand docking for drug discovery. We also discuss current approaches to address major challenges of docking. Key Words: chemoinformatics, computer-aided drug design, drug discovery, structure-activity relationships. Acoplamiento Molecular: Avances Recientes y Retos RESUMEN El acoplamiento molecular automatizado tiene como objetivo proponer un modelo de unión entre dos moléculas. Este método ha sido útil en química farmacéutica y en el descubrimiento de nuevos fármacos por medio del entendimiento de las fuerzas de interacción involucradas en el reconocimiento molecular.