The Variation of Genome Sites Associated with Severe Covid-19 Across Populations: the Worldwide and National Patterns
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Climate Warming Impacts on Distributions of Scots Pine (Pinus Sylvestris L.) Seed Zones and Seed Mass Across Russia in the 21St Century
Article Climate Warming Impacts on Distributions of Scots Pine (Pinus sylvestris L.) Seed Zones and Seed Mass across Russia in the 21st Century Elena I. Parfenova 1, Nina A. Kuzmina 2, Sergey R. Kuzmin 2 and Nadezhda M. Tchebakova 1,* 1 Laboratory of Forest Monitoring, Sukachev Institute of Forest FRC KSC SB RAS, 660036 Krasnoyarsk, Russia; [email protected] 2 Laboratory of Forest Genetics and Breeding, Sukachev Institute of Forest FRC KSC SB RAS, 660036 Krasnoyarsk, Russia; [email protected] (N.A.K.); [email protected] (S.R.K.) * Correspondence: [email protected] Abstract: Research highlights: We investigated bioclimatic relationships between Scots pine seed mass and seed zones/climatypes across its range in Russia using extensive published data to predict seed zones and seed mass distributions in a changing climate and to reveal ecological and genetic components in the seed mass variation using our 40-year common garden trial data. Introduction: seed productivity issues of the major Siberian conifers in Asian Russia become especially relevant nowadays in order to compensate for significant forest losses due to various disturbances during the 20th and current centuries. Our goals were to construct bioclimatic models that predict the seed mass of major Siberian conifers (Scots pine, one of the major Siberian conifers) in a warming climate during the current century. Methods: Multi-year seed mass data were derived from the literature and were collected during field work. Climate data (January and July data and annual precipitation) were Citation: Parfenova, E.I.; Kuzmina, derived from published reference books on climate and climatic websites. -
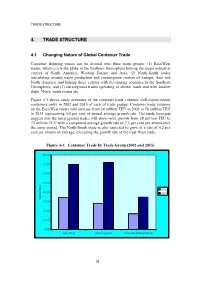
4. Trade Structure
TRADE STRUCTURE 4. TRADE STRUCTURE 4.1 Changing Nature of Global Container Trade Container shipping routes can be divided into three main groups: (1) East-West trades, which circle the globe in the Northern Hemisphere linking the major industrial centres of North America, Western Europe and Asia; (2) North-South trades articulating around major production and consumption centres of Europe, Asia and North America, and linking these centres with developing countries in the Southern Hemisphere; and (3) intraregional trades operating in shorter hauls and with smaller ships. North–south routes are Figure 4-1 shows study estimates of the container trade volumes (full export/import containers only) in 2002 and 2015 of each of trade groups. Container trade volumes on the East-West routes will increase from 34 million TEU in 2002 to 70 million TEU in 2015 representing 5.8 per cent of annual average growth rate. The study forecasts suggest that the intraregional trades will show solid growth from 28 million TEU to 72 million TEU with a compound average growth rate of 7.5 per cent per annum over the same period. The North-South trade is also expected to grow at a rate of 6.2 per cent per annum on average, exceeding the growth rate of the East-West trade. Figure 4-1: Container Trade by Trade Group (2002 and 2015) 80.00 70.00 60.00 50.00 2002 40.00 2015 Million TEU 30.00 20.00 10.00 0.00 East-West Intra-Regional North-South/South-South 32 TRADE STRUCTURE 4.2 Asia - North America The biggest deep sea liner route is the trans-Pacific trade between Asia and North America, representing 14.5 million TEU in 2002, equivalent to 43 per cent of the total East-West trade and 19 per cent of the world total. -

North and Central Asia FAO-Unesco Soil Tnap of the World 1 : 5 000 000 Volume VIII North and Central Asia FAO - Unesco Soil Map of the World
FAO-Unesco S oilmap of the 'world 1:5 000 000 Volume VII North and Central Asia FAO-Unesco Soil tnap of the world 1 : 5 000 000 Volume VIII North and Central Asia FAO - Unesco Soil map of the world Volume I Legend Volume II North America Volume III Mexico and Central America Volume IV South America Volume V Europe Volume VI Africa Volume VII South Asia Volume VIIINorth and Central Asia Volume IX Southeast Asia Volume X Australasia FOOD AND AGRICULTURE ORGANIZATION OF THE UNITED NATIONS UNITED NATIONS EDUCATIONAL, SCIENTIFIC AND CULTURAL ORGANIZATION FAO-Unesco Soilmap of the world 1: 5 000 000 Volume VIII North and Central Asia Prepared by the Food and Agriculture Organization of the United Nations Unesco-Paris 1978 The designations employed and the presentation of material in this publication do not irnply the expression of any opinion whatsoever on the part of the Food and Agriculture Organization of the United Nations or of the United Nations Educa- tional, Scientific and Cultural Organization con- cerning the legal status of any country, territory, city or area or of its authorities, or concerning the delirnitation of its frontiers or boundaries. Printed by Tipolitografia F. Failli, Rome, for the Food and Agriculture Organization of the United Nations and the United Nations Educational, Scientific and Cultural Organization Published in 1978 by the United Nations Educational, Scientific and Cultural Organization Place de Fontenoy, 75700 Paris C) FAO/Unesco 1978 ISBN 92-3-101345-9 Printed in Italy PREFACE The project for a joint FAO/Unesco Soil Map of vested with the responsibility of compiling the techni- the World was undertaken following a recommenda- cal information, correlating the studies and drafting tion of the International Society of Soil Science. -

Country Profile: Russia Note: Representative
Country Profile: Russia Introduction Russia, the world’s largest nation, borders European and Asian countries as well as the Pacific and Arctic oceans. Its landscape ranges from tundra and forests to subtropical beaches. It’s famous for novelists Tolstoy and Dostoevsky, plus the Bolshoi and Mariinsky ballet companies. St. Petersburg, founded by legendary Russian leader Peter the Great, features the baroque Winter Palace, now housing part of the Hermitage Museum’s art collection. Extending across the entirety of northern Asia and much of Eastern Europe, Russia spans eleven time zones and incorporates a wide range of environments and landforms. From north west to southeast, Russia shares land borders with Norway, Finland, Estonia, Latvia, Lithuania and Poland (both with Kaliningrad Oblast), Belarus, Ukraine, Georgia, Azerbaijan, Kazakhstan, China,Mongolia, and North Korea. It shares maritime borders with Japan by the Sea of Okhotsk and the U.S. state of Alaska across the Bering Strait. Note: Representative Map Population The total population of Russia during 2015 was 142,423,773. Russia's population density is 8.4 people per square kilometre (22 per square mile), making it one of the most sparsely populated countries in the world. The population is most dense in the European part of the country, with milder climate, centering on Moscow and Saint Petersburg. 74% of the population is urban, making Russia a highly urbanized country. Russia is the only country 1 Country Profile: Russia in the world where more people are moving from cities to rural areas, with a de- urbanisation rate of 0.2% in 2011, and it has been deurbanising since the mid-2000s. -

Defining Territories and Empires: from Mongol Ulus to Russian Siberia1200-1800 Stephen Kotkin
Defining Territories and Empires: from Mongol Ulus to Russian Siberia1200-1800 Stephen Kotkin (Princeton University) Copyright (c) 1996 by the Slavic Research Center All rights reserved. The Russian empire's eventual displacement of the thirteenth-century Mongol ulus in Eurasia seems self-evident. The overthrow of the foreign yoke, defeat of various khanates, and conquest of Siberia constitute core aspects of the narratives on the formation of Russia's identity and political institutions. To those who disavow the Mongol influence, the Byzantine tradition serves as a counterweight. But the geopolitical turnabout is not a matter of dispute. Where Chingis Khan and his many descendants once held sway, the Riurikids (succeeded by the Romanovs) moved in. *1 Rather than the shortlived but ramified Mongol hegemony, which was mostly limited to the middle and southern parts of Eurasia, longterm overviews of the lands that became known as Siberia, or of its various subregions, typically begin with a chapter on "pre-history," which extends from the paleolithic to the moment of Russian arrival in the late sixteenth, early seventeenth centuries. *2 The goal is usually to enable the reader to understand what "human material" the Russians found and what "progress" was then achieved. Inherent in the narratives -- however sympathetic they may or may not be to the native peoples -- are assumptions about the historical advance deriving from the Russian arrival and socio-economic transformation. In short, the narratives are involved in legitimating Russia's conquest without any notion of alternatives. Of course, history can also be used to show that what seems natural did not exist forever but came into being; to reveal that there were other modes of existence, which were either pushed aside or folded into what then came to seem irreversible. -

History and Social Science
Nashoba Regional School District HISTORY AND SOCIAL SCIENCE Standards and Benchmarks Grade 7 Nashoba Regional School District History and Social Science Standards and Benchmarks, 2007-2008. Work in this document is based upon the standards outlined in the Massachusetts History and Social Science Framework, August 2003. History and Social Studies by Grade Level Grade 7 Massachusetts Curriculum Frameworks (August 2003) Overarching Theme The study of world geography includes 5 majors themes: location, place, human interaction with the environment, movement, and regions. Grade Seven Focus: Foundations of Geography Concepts and Skills to be addressed: Students will be able to identify and interpret different kinds of maps, charts, graphs. They will use geographic and demographic terms correctly and use an atlas. They will understand what a nation is and the different international organizations. They will understand basic economic concepts such as supply and demand, economic systems, trade and the effect of these on the standard of living. Big Idea: A nation’s standard of living is impacted by its economic system, its government and geographical characteristics. Essential Questions: In what ways does location affect way of life? What qualities make a nation and how do nations interact? How does the economic system affect standard of living? By the end of SEVENTH Grade, students should be able to answer the Essential Questions above and apply knowledge and concepts attained to be able to: History and Geography • Use map and globe skills learned in pre-kindergarten to grade five to interpret different kinds of projections, as well as topographic, landform, political, population, and climate maps. -

Global Migration and Regionalization, 1840-1940
GLOBAL MIGRATION AND REGIONALIZATION, 1840-1940 Paper for conference on Mapping Global Inequalities Santa Cruz, California December 13-14, 2007 Adam McKeown Associate Professor of History Columbia University [email protected] The mass migrations of the nineteenth and early twentieth centuries were a global phenomenon. From the North Atlantic to the South Pacific, hardly any corner of the earth was untouched by migration. These migrations similar in quantity and organization, and all linked through the processes of globalization: the peopling of frontiers, new transportation technologies, the production and processing of material for modern industry, the shipment and marketing of finished goods, and the production of food, shelter and clothing for people who worked in those industrial and distribution networks. It was a truly global process. Yet, the processes and cycles of migration grew increasingly integrated across the globe, the actual patterns and directions of migration grew more regionally segregated. These segregated regions experienced different patterns 2 of development and growth associated with migration. Moreover, this segregation helped to erase many of the non-Atlantic migrations from the historical memory, thus helping to obscure inequalities that were created as part of historical globalization by depicting certain parts of the world as having been outside of globalization. Most histories have recounted the age of mass migration as a transatlantic age. When migrations beyond the Atlantic are remembered at all, it is usually as a limited number of indentured laborers pressed into the service of Europeans. This historical memory is not random. It is the foundation of the broadest of global identities: East and West, North and South, First and Third Worlds, and the liberal and illiberal regions of the modern international system. -

Mitochondrial Genome Diversity in the Central Siberian Plateau with Particular
bioRxiv preprint doi: https://doi.org/10.1101/656181; this version posted May 31, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Mitochondrial Genome Diversity in the Central Siberian Plateau with Particular Reference to Prehistory of Northernmost Eurasia S. V. Dryomov*,1, A. M. Nazhmidenova*,1, E. B. Starikovskaya*,1, S. A. Shalaurova1, N. Rohland2, S. Mallick2,3,4, R. Bernardos2, A. P. Derevianko5, D. Reich2,3,4, R. I. Sukernik1. 1 Laboratory of Human Molecular Genetics, Institute of Molecular and Cellular Biology, SBRAS, Novosibirsk, Russian Federation 2 Department of Genetics, Harvard Medical School, Boston, MA 02115, USA 3 Broad Institute of Harvard and MIT, Cambridge, MA 02142, USA 4 Howard Hughes Medical Institute, Harvard Medical School, Boston, MA 02115, USA 5 Institute of Archaeology and Ethnography, SBRAS, Novosibirsk, Russian Federation Corresponding author: Rem Sukernik ([email protected]) * These authors contributed equally to this work. bioRxiv preprint doi: https://doi.org/10.1101/656181; this version posted May 31, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Abstract The Central Siberian Plateau was last geographic area in Eurasia to become habitable by modern humans after the Last Glacial Maximum (LGM). -

Regional Fact Sheet
SIXTH ASSESSMENT REPORT Working Group I – The Physical Science Basis Regional fact sheet - Asia Common regional changes • The observed mean surface temperature increase has clearly emerged out of the range of internal variability compared to 1850-1900. Heat extremes have increased while cold extremes have decreased, and these trends will continue over the coming decades (high confidence). • Marine heatwaves will continue to increase (high confidence). • Fire weather seasons will lengthen and intensify, particularly in North Asia regions (medium confidence). • Average and heavy precipitation will increase over much of Asia (high to medium confidence). • Mean surface wind speeds have decreased (high confidence) and will continue to decrease in central and northern parts of Asia (medium confidence). • Glaciers are declining and permafrost is thawing. Seasonal snow duration, glacial mass, and permafrost area will decline further by the mid-21st century (high confidence). • Glacier runoff in the Asian high mountains will increase up to mid-21st century (medium confidence), and subsequently runoff may decrease due to the loss of glacier storage. • Relative sea level around Asia has increased faster than global average, with coastal area loss and shoreline retreat. Regional-mean sea level will continue to rise (high confidence). Annual mean temperature DJF Total precipitation JJA Total precipitation Max. temperature over 35oC Projections at 1.5ºC, 2ºC, and 4ºC global warming Changes relative to the 1850–1900 in annual mean surface temperature, total precipitation (Dec– Feb, DJF), and total precipitation (Jun– Aug, JJA). Last column shows the total number of days per year with maximum temperature exceeding 35ºC. Results expanded in the Interactive Atlas (active links) Asian Monsoons • The South and Southeast Asian monsoon has weakened in the second half of the 20th century (high confidence). -

List of Countries Per Region.Xlsx
Country Council Region Afghanistan South Asia Albania Eastern & Central Europe Algeria Africa American Samoa Australia, New Zealand & Polynesia Andorra Mediterranean Europe Angola Africa Antigua and Barbuda North America Argentina Latin America Armenia NIS & Russia Aruba North America Australia Australia, New Zealand & Polynesia Austria Continental Europe Azerbaijan NIS & Russia The Bahamas North America Bahrain Middle East Bangladesh South Asia Barbados North America Belarus NIS & Russia Belgium Continental Europe Belize Latin America Benin Africa Bermuda North America Bhutan South Asia Bolivia Latin America Bosnia and Herzegovina Eastern & Central Europe Botswana Africa Brazil Latin America Brunei South East Asia Bulgaria Eastern & Central Europe Burkina Faso Africa Burundi Africa Cabo Verde Africa Cambodia South East Asia Cameroon Africa Canada North America Cayman Islands North America Central African Republic Africa Chad Africa Channel Islands Continental Europe Chile Latin America China East Asia Colombia Latin America Comoros Africa Congo Africa Costa Rica Latin America Country Council Region Côte D'Ivoire Africa Croatia Eastern & Central Europe Cuba Latin America Curaçao Latin America Cyprus Eastern & Central Europe Czech Republic Eastern & Central Europe Dem. People's Rep. Korea North Asia Dem. Rep. Congo Africa Denmark Scandinavia Djibouti Africa Dominica Latin America Dominican Republic Latin America Ecuador Latin America Egypt Africa El Salvador Latin America Equatorial Guinea Africa Eritrea Africa Estonia Eastern & Central -

Seasonality, Maintenance, and Climate Impact on North America
Clim Dyn DOI 10.1007/s00382-017-3734-6 The Asian–Bering–North American teleconnection: seasonality, maintenance, and climate impact on North America Bin Yu1 · H. Lin2 · Z. W. Wu3 · W. J. Merryfield4 Received: 23 January 2017 / Accepted: 12 May 2017 © The Author(s) 2017. This article is an open access publication Abstract The Asian–Bering–North American (ABNA) be traced back to the preceding season, which provides a teleconnection index is constructed from the normalized predictability source for this teleconnection and for North 500-hPa geopotential field by excluding the Pacific–North American temperature variability. The ABNA associated American pattern contribution. The ABNA pattern fea- energy budget is dominated by surface longwave radiation tures a zonally elongated wavetrain originating from North anomalies year-round, with the temperature anomalies sup- Asia and flowing downstream across Bering Sea and Strait ported by anomalous downward longwave radiation and towards North America. The large-scale teleconnection damped by upward longwave radiation at the surface. is a year-round phenomenon that displays strong season- ality with the peak variability in winter. North American surface temperature and temperature extremes, including 1 Introduction warm days and nights as well as cold days and nights, are significantly controlled by this teleconnection. The ABNA The impact of the tropical sea surface temperature (SST) pattern has an equivalent barotropic structure in the tropo- anomaly, in particular the El Niño–Southern Oscilla- sphere and is supported by synoptic-scale eddy forcing in tion (ENSO) variability, on North American climate has the upper troposphere. Its associated sea surface tempera- been extensively investigated (e.g., Ropelewski and Halp- ture anomalies exhibit a horseshoe-shaped structure in the ert 1986; Trenberth et al. -

Challenges for the Russian Far East in the Asia-Pacific Region
Integration or Disintegration: Challenges for the Russian Far East in the Asia-Pacific Region Tamara Troyakova and Elizabeth Wishnick The disintegration of economic links within the Russian Federa- tion has propelled the regions comprising the Russian Far East to find new markets in Asia, but, ironically, the very weakness of the Russian state also has proved to be the greatest obstacle to the economic inte- gration of these regions with the Pacific Rim economy. Russia’s flawed mechanisms for coordinating center-regional relations and poorly developed regional institutions, have limited the ability of the Russian Far East to promote economic relations with Asian neighbors. In the past three years President Vladimir Putin has taken steps to restructure center-regional relations in hope of creating a more effec- tive state. We examine the consequences of these reforms both for Russia's future political development and for the economic integration of the Russian Far East in Northeast Asia. This paper examines the twin challenges confronting the Russian Far East: 1) economic integration in the Asia-Pacific economy, a region that has been emblematic of robust trade but weakly institutionalized economic linkages, and 2) political disintegration within Russia, resulting from ineffective patterns of center-regional relations, crime, and corruption. Particular attention is directed to trade with China, Japan, the United States and South Korea, investment in transportation and energy pro- jects, and labor cooperation with China and North Korea. Regionalism, Economic Integration, and the State Initial faith in the ability of the Russian Far East to become a part of Asia’s dynamic economy coincided with the boom in intra-Asian trade and investment in the first half of the 1990s.