Reactions Involving Additions And/Or Eliminations Addition Reactions 10.2 Hydration of Carbonyl Structures
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Chemistry Department 32.Pdf (10.36Mb)
THE FABRICATION OF NANOMETRIC METAL SULFIDES FROM XANTHATE PRECURSORS By ALAN PIQUETTE Bachelor of Arts Western State College of Colorado Gunnison, Colorado 2002 Submitted to the Faculty of the Graduate College of the Oklahoma State University In partial fulfillment of The requirements for The Degree of DOCTOR OF PHILOSOPHY December, 2007 THE FABRICATION OF NANOMETRIC METAL SULFIDES FROM XANTHATE PRECURSORS Thesis Approved: _______________ __Allen Apblett___ ______________ Thesis Advisor ________________ Nicholas Materer_____ __________ _____________ __LeGrande Slaughter___ __________ _______________ ____Jim Smay___ _______________ _____________ __A. Gordon Emslie___ ____________ Dean of the Graduate College ACKNOWLEDGEMENTS It is with my utmost sincere appreciation that I acknowledge my thesis advisor, Dr. Allen W. Apblett, for his support, guidance, and motivation. He provided a research environment that was both friendly and challenging, which made my years in his group enjoyable and rewarding. His broad range of knowledge was something of which I was happy to take advantage. His dedication to teaching and research is something that will be a positive influence on me for the rest of my scientific career. I would like to express gratitude to my committee members: Dr. Nicholas Materer, Dr. LeGrande Slaughter, and Dr. Jim Smay, for their assistance, advice, guidance, and support throughout the years. I am deeply grateful to all my colleagues and friends in the chemistry department. I would specifically like to thank Sulaiman Al-Fadul, Mohammed Al-Hazmi, Zeid Al- Othmann, Mohamed Chehbouni, Satish Kuriyavar, and Tarek Trad for showing me the ropes, for their valuable discussions, continuous encouragement, and for all the help they extended during the course of my stay in the Apblett Group. -

I the Synthesis of Papyracillic Acids: Application of the Tandem Chain
University of New Hampshire University of New Hampshire Scholars' Repository Doctoral Dissertations Student Scholarship Winter 2011 I The synthesis of papyracillic acids: Application of the tandem chain extension-acylation reaction II Diastereoselective synthesis of beta-methyl-gamma-keto esters through the utility of an L- serine auxiliary Jennifer R. Mazzone University of New Hampshire, Durham Follow this and additional works at: https://scholars.unh.edu/dissertation Recommended Citation Mazzone, Jennifer R., "I The synthesis of papyracillic acids: Application of the tandem chain extension- acylation reaction II Diastereoselective synthesis of beta-methyl-gamma-keto esters through the utility of an L-serine auxiliary" (2011). Doctoral Dissertations. 644. https://scholars.unh.edu/dissertation/644 This Dissertation is brought to you for free and open access by the Student Scholarship at University of New Hampshire Scholars' Repository. It has been accepted for inclusion in Doctoral Dissertations by an authorized administrator of University of New Hampshire Scholars' Repository. For more information, please contact [email protected]. I. THE SYNTHESES OF PAPYRACILLIC ACIDS: APPLICATION OF THE TANDEM CHAIN EXTENSION-ACYLATION REACTION II. DIASTEREOSELECTIVE SYNTHESIS OF p-METHYL-y-KETO ESTERS THROUGH THE UTILITY OF AN L-SERINE AUXILIARY BY Jennifer R. Mazzone B.A., Assumption College, 2006 DISSERTATION Submitted to the University of New Hampshire in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy in Chemistry December, 2011 UMI Number: 3500790 All rights reserved INFORMATION TO ALL USERS The quality of this reproduction is dependent upon the quality of the copy submitted. In the unlikely event that the author did not send a complete manuscript and there are missing pages, these will be noted. -

Organic Chemistry
Wisebridge Learning Systems Organic Chemistry Reaction Mechanisms Pocket-Book WLS www.wisebridgelearning.com © 2006 J S Wetzel LEARNING STRATEGIES CONTENTS ● The key to building intuition is to develop the habit ALKANES of asking how each particular mechanism reflects Thermal Cracking - Pyrolysis . 1 general principles. Look for the concepts behind Combustion . 1 the chemistry to make organic chemistry more co- Free Radical Halogenation. 2 herent and rewarding. ALKENES Electrophilic Addition of HX to Alkenes . 3 ● Acid Catalyzed Hydration of Alkenes . 4 Exothermic reactions tend to follow pathways Electrophilic Addition of Halogens to Alkenes . 5 where like charges can separate or where un- Halohydrin Formation . 6 like charges can come together. When reading Free Radical Addition of HX to Alkenes . 7 organic chemistry mechanisms, keep the elec- Catalytic Hydrogenation of Alkenes. 8 tronegativities of the elements and their valence Oxidation of Alkenes to Vicinal Diols. 9 electron configurations always in your mind. Try Oxidative Cleavage of Alkenes . 10 to nterpret electron movement in terms of energy Ozonolysis of Alkenes . 10 Allylic Halogenation . 11 to make the reactions easier to understand and Oxymercuration-Demercuration . 13 remember. Hydroboration of Alkenes . 14 ALKYNES ● For MCAT preparation, pay special attention to Electrophilic Addition of HX to Alkynes . 15 Hydration of Alkynes. 15 reactions where the product hinges on regio- Free Radical Addition of HX to Alkynes . 16 and stereo-selectivity and reactions involving Electrophilic Halogenation of Alkynes. 16 resonant intermediates, which are special favor- Hydroboration of Alkynes . 17 ites of the test-writers. Catalytic Hydrogenation of Alkynes. 17 Reduction of Alkynes with Alkali Metal/Ammonia . 18 Formation and Use of Acetylide Anion Nucleophiles . -

Nucleophilic Aromatic Substitution
NUCLEOPHILIC AROMATIC SUBSTITUTION Ms. Prerana Sanas M.Pharm (Pharmceutical Chemistry) Asst.Professor, NCRD’s Sterling Institute of Pharmacy Nerul Navi Mumbai Nucleophilic aromatic substitution results in the substitution of a halogen X on a benzene ring by a nucleophile (:Nu– ). Aryl halides undergo a limited number of substitution reactions with strong nucleophiles. NAS occurs by two mechanisms i) Bimoleccular displacement (Addition –Elimination) ii) Benzyne Formation( Elimination –Addition) 7/5/2019 Ms.Prerana Sanas 2 Bimolecular displacement (Addition – Elimination) Aryl halides with strong electron-withdrawing groups (such as NO2) on the ortho or para positions react with nucleophiles to afford substitution products. For example, treatment of p-chloronitrobenzene with hydroxide (– OH) affords p-nitrophenol by replacement of Cl by OH. Nucleophilic aromatic substitution occurs with a variety of strong nucleophiles, including – OH, – OR, – NH2, – SR, and in some cases, neutral nucleophiles such as NH3 and RNH2 . 7/5/2019 Ms.Prerana Sanas 3 Mechanism…… The mechanism of these reactions has two steps: Step i) Addition of the nucleophile (:Nu– ) forms a resonance-stabilized carbanion with a new C – Nu bond—three resonance structures can be drawn. • Step [1] is rate-determining since the aromaticity of the benzene ring is lost. In Step ii) loss of the leaving group re-forms the aromatic ring. This step is fast because the aromaticity of the benzene ring is restored. 7/5/2019 Ms.Prerana Sanas 4 Factors affecting Bimolecular displacement Increasing the number of electron-withdrawing groups increases the reactivity of the aryl halide. Electron-withdrawing groups stabilize the intermediate carbanion, and by the Hammond postulate, lower the energy of the transition state that forms it. -

Chemical Kinetics HW1 (Kahn, 2010)
Chemical Kinetics HW1 (Kahn, 2010) Question 1. (6 pts) A reaction with stoichiometry A = P + 2Q was studied by monitoring the concentration of the reactant A as a function of time for eighteen minutes. The concentration determination method had a maximum error of 6 M. The following concentration profile was observed: Time (min) Conc (mM) 1 0.9850 2 0.8571 3 0.7482 4 0.6549 5 0.5885 6 0.5183 7 0.4667 8 0.4281 9 0.3864 10 0.3557 11 0.3259 12 0.3037 13 0.2706 14 0.2486 15 0.2355 16 0.2188 17 0.2111 18 0.1930 Determine the reaction order and calculate the rate constant for decomposition of A. What can be said about the mechanism or molecularity of this reaction? Question 2. (4 pts) Solve problem 2 on pg 31 in your textbook (House) using both linear and non-linear regression. Provide standard errors for the rate constant and half-life based on linear and non-linear fits. Below is the data set for your convenience: dataA = {{0, 0.5}, {10, 0.443}, {20,0.395}, {30,0.348}, {40,0.310}, {50,0.274}, {60,0.24}, {70,0.212}, {80,0.190}, {90,0.171}, {100,0.164}} Question 3. (3 pts) Solve problem 3 on pg 32 in your textbook (House). Question 4. (7 pts) The authors of the paper “Microsecond Folding of the Cold Shock Protein Measured by a Pressure-Jump Technique” suggest that the activated state of folding of CspB follows Hammond- type behavior. -

Lecture 5 Diastereoselective Addition Into Carbonyl Compounds
Lecture 5 Diastereoselective Addition into Carbonyl Compounds Containing α-Stereogenic Centres Learning outcomes: by the end of this lecture, and after answering the associated problems, you will be able to: 1. use the Felkin-Anh T.S. to predict the stereochemical outcome of reactions carried out on carbonyl compounds that possess an α-stereogenic centre; 2. rationalise the preferential adoption of a Felkin-Anh T.S. in nucleophilic addition reactions on steric and stereoelectronic grounds; 3. understand how the presence of an α-electron-withdrawing substituent affects the Felkin-Anh T.S.; 4. use the Felkin-Anh T.S. to prepare 1,2-syn diols from α-alkoxy ketones; 5. use the Cram chelation model to prepare 1,2-anti diols from α-alkoxy ketones. Stereoselective Addition of Nucleophiles into Ketones and Aldehydes containing α- Stereogenic Centres The addition of a nucleophile into a chiral ketone or aldehyde provides diastereoisomers. When the stereogenic centres in the substrate are close to the reacting carbonyl group (e.g. 1,2-disposed), then it is often possible to exploit this stereochemical information to control the stereoselectivity of the addition reaction. This method for controlling the stereochemical outcome of a reaction is known as substrate control. A number of models have been developed for predicting the stereochemical outcome of this type of reaction. Felkin-Anh Model Consider a carbonyl compound containing an α-stereogenic centre in which the three substituents at the α-site are well differentiated in size: O HO Nu Nu OH RL Nu RL RL R R R S M S M S RM R R R R R RS = small substituent RM = medium-sized substituent RL = large substituent Of the two diastereoisomeric alcohol addition products, one will be formed to a greater extent than the other. -

2 Reactions Observed with Alkanes Do Not Occur with Aromatic Compounds 2 (SN2 Reactions Never Occur on Sp Hybridized Carbons!)
Reactions of Aromatic Compounds Aromatic compounds are stabilized by this “aromatic stabilization” energy Due to this stabilization, normal SN2 reactions observed with alkanes do not occur with aromatic compounds 2 (SN2 reactions never occur on sp hybridized carbons!) In addition, the double bonds of the aromatic group do not behave similar to alkene reactions Aromatic Substitution While aromatic compounds do not react through addition reactions seen earlier Br Br Br2 Br2 FeBr3 Br With an appropriate catalyst, benzene will react with bromine The product is a substitution, not an addition (the bromine has substituted for a hydrogen) The product is still aromatic Electrophilic Aromatic Substitution Aromatic compounds react through a unique substitution type reaction Initially an electrophile reacts with the aromatic compound to generate an arenium ion (also called sigma complex) The arenium ion has lost aromatic stabilization (one of the carbons of the ring no longer has a conjugated p orbital) Electrophilic Aromatic Substitution In a second step, the arenium ion loses a proton to regenerate the aromatic stabilization The product is thus a substitution (the electrophile has substituted for a hydrogen) and is called an Electrophilic Aromatic Substitution Energy Profile Transition states Transition states Intermediate Potential E energy H Starting material Products E Reaction Coordinate The rate-limiting step is therefore the formation of the arenium ion The properties of this arenium ion therefore control electrophilic aromatic substitutions (just like any reaction consider the stability of the intermediate formed in the rate limiting step) 1) The rate will be faster for anything that stabilizes the arenium ion 2) The regiochemistry will be controlled by the stability of the arenium ion The properties of the arenium ion will predict the outcome of electrophilic aromatic substitution chemistry Bromination To brominate an aromatic ring need to generate an electrophilic source of bromine In practice typically add a Lewis acid (e.g. -

Ketenes 25/01/2014 Part 1
Baran Group Meeting Hai Dao Ketenes 25/01/2014 Part 1. Introduction Ph Ph n H Pr3N C A brief history Cl C Ph + nPr NHCl Ph O 3 1828: Synthesis of urea = the starting point of modern organic chemistry. O 1901: Wedekind's proposal for the formation of ketene equivalent (confirmed by Staudinger 1911) Wedekind's proposal (1901) 1902: Wolff rearrangement, Wolff, L. Liebigs Ann. Chem. 1902, 325, 129. 2 Wolff adopt a ketene structure in 1912. R 2 hν R R2 1905: First synthesis and characterization of a ketene: in an efford to synthesize radical 2, 1 ROH R C Staudinger has synthesized diphenylketene 3, Staudinger, H. et al., Chem. Ber. 1905, 1735. N2 1 RO CH or Δ C R C R1 1907-8: synthesis and dicussion about structure of the parent ketene, Wilsmore, O O J. Am. Chem. Soc. 1907, 1938; Wilsmore and Stewart Chem. Ber. 1908, 1025; Staudinger and Wolff rearrangement (1902) O Klever Chem. Ber. 1908, 1516. Ph Ph Cl Zn Ph O hot Pt wire Zn Br Cl Cl CH CH2 Ph C C vs. C Br C Ph Ph HO O O O O O O O 1 3 (isolated) 2 Wilsmore's synthesis and proposal (1907-8) Staudinger's synthesis and proposal (1908) wanted to make Staudinger's discovery (1905) Latest books: ketene (Tidwell, 1995), ketene II (Tidwell, 2006), Science of Synthesis, Vol. 23 (2006); Latest review: new direactions in ketene chemistry: the land of opportunity (Tidwell et al., Eur. J. Org. Chem. 2012, 1081). Search for ketenes, Google gave 406,000 (vs. -

Reaction Mechanism Lecture 3 : Nucleophilic And
Module 10 : Reaction mechanism Lecture 3 : Nucleophilic and Electrophilic Addition and Abstraction Objectives In this lecture you will learn the following Ligand activation by metal that leads to a direct external attack at the ligand. Nucleophilic addition and nucleophilic abstraction reactions. Electrophilic addition and electrophilic abstraction reactions. The nucleophilic and electrophilic substitution and abstraction reactions can be viewed as ways of activation of substrates to allow an external reagent to directly attack the metal activated ligand without requiring prior binding of the external reagent to the metal. The attacking reagent may be a nucleophile or an electrophile. The nucleophilic attack of the external reagent is favored if the LnM fragment is a poor π−base and a good σ−acid i.e., when the complex is cationic and/or when the other metal bound ligands are electron withdrawing such that the ligand getting activated gets depleted of electron density and can undergo an external attack by a nucleophile Nu−, like LiMe or OH−. The attack of the nucleophiles may result in the formation of a bond between the nucleophiles and the activated unsaturated substrate, in which case it is called nucleophilic addition, or may result in an abstraction of a part or the whole of the activated ligand, in which case it is called the nucleophilic abstraction. The nucleophilic addition and the abstraction reactions are discussed below. Nucleophilic addition An example of a nucleophilic addition reaction is shown below. Carbon monoxide (CO) as a ligand can undergo nucleophilic attack when bound to a metal center of poor π−basicity, as the carbon center of the CO ligand is electron deficient owing to the ligand to metal σ−donation not being fully compensated by the metal to ligand π−back donation. -
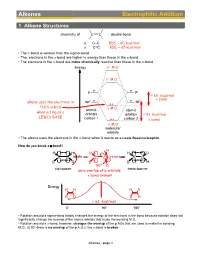
Alkenes Electrophilic Addition
Alkenes Electrophilic Addition 1 Alkene Structures chemistry of C C double bond σ C–C BDE ~ 80 kcal/mol π C=C BDE ~ 65 kcal/mol • The p-bond is weaker than the sigma-bond • The, electrons in the p-bond are higher in energy than those in the s-bond • The electrons in the p-bond are more chemically reactive than those in the s-bond Energy σ∗ M.O. π∗ M.O. p p ~ 65 kcal/mol 2 π bond alkene uses the electrons in sp2 sp THIS orbital π M.O. atomic atomic when acting as a orbitals orbitals ~ 81 kcal/mol LEWIS BASE carbon 1 carbon 2 σ bond σ M.O. molecular orbitals • The alkene uses the electrons in the p-bond when it reacts as a Lewis Base/nucleophile How do you break a p-bond? H H H H H D C C rotate C C rotate C C D D D D H 90° D cis-isomer trans-isomer zero overlap of p orbitals: π bond broken! Energy H H H D D D D H ~ 63 kcal/mol 0° 90° 180° • Rotation around a sigma-bond hardly changes the energy of the electrons in the bond because rotation does not significantly change the overlap of the atomic orbitals that make the bonding M.O. • Rotation around a p-bond, however, changes the overlap of the p AOs that are used to make the bonding M.O., at 90° there is no overlap of the p A.O.s, the p-bond is broken Alkenes : page 1 Distinguishing isomers trans- cis- • By now we are very familiar with cis- and trans-stereoisomers (diastereomers) • But what about, the following two structures, they can NOT be assigned as cis- or trans-, yet they are definitely stereoisomers (diastereomers), the directions in which their atoms point in space are different cis- / trans- Br Br We Need a different system to distinguish stereoisomers for C=C double bonds: Use Z/E notation. -

Subject Index
455 Subject Index Aminohydroxylation, 364 a-Aminoketone, 281 4-Aminophenol, 18 A Aminothiophene, 158 Abnormal Claisen rearrangement, 1 a-Aminothiophenols, 184 Acrolein, 378 Ammonium ylide, 383 2-(Acylamino)-toluenes, 245 Angeli-Rimini hydroxamic acid Acylation, 100, 145, 200 synthesis, 9 Acyl azides, 98 ~-Anomer, 225 Acylium ion, 145, 149, 175, 177 Anomeric center, 211 a-Acyloxycarboxamides, 298 Anomeric effect, 135 a-Acyloxyketones, 17 ANRORC mechanism, 10 a-Acyloxythioethers, 327 Anthracenes, 51 Acyl transfer, 17, 42, 228, 298, 305, Anti-Markovnikov addition, 219 327,345 Amdt-Eistert homologation, 11 AIBN, 22, 23, 415 Aryl-acetylene, 66 Alder ene reaction, 2 Arylation, 253 Alder's endo rule, Ill 0-Aryliminoethers, 67 Aldol condensation, 3, 14, 26, 34, 2-Arylindoles, 38 69, 130, 147, 172, 305, Aryl migration, 31 340,396,412 Autoxidation, 69, 115, 118 Aldosylamine, 8 Auwers reaction, 13 Alkyl migration, 16, 132, 315, 443 Axial, 347 Alkylation, 144, 145 Azalactone, I 00 N-Aikylation, 162 Azides, 125, 330 Alkylidene carbene, 151 Azirine, 6, 7, 281 Allan-Robinson reaction, 4, 228 Azulene, 310 Allene, 119 1t-Allyl complex, 414 B Allylation, 213, 414 Baeyer-Drewson Allylstannane, 213 indigo synthesis, 14 Allylsilanes, 349 Baeyer-Villiger oxidation, 16, 53 Alper carbonylation, 6 Baker-Venkataraman Alpine-borane®, 262 rearrangement, 17 Aluminum phenolate, 149 Balz-Schiemann reaction, 354 Amadori rearrangement, 8 Bamberger rearrangement, 18 Amide acetal, 74 Bamford-Stevens reaction, 19 Amides, 28, 67,276,339, 356 Bargellini reaction, 20 Amidine, -

Aromatic Nucleophilic Substitution Reaction
Aromatic Nucleophilic Substitution Reaction DR. RAJENDRA R TAYADE ASSISTANT PROFESSOR DEPARTMENT OF CHEMISTRY INSTITUTE OF SCIENCE, NAGPUR Principles There are four principal mechanisms for aromatic nucleophilic substitution which are similar to that of aliphatic nucleophilic substitution. (SN1, SN2, SNi, SET Mechanism) 1. SNAr Mechanism- addition / elimination CF3, CN, CHO, COR, COOH, Br, Cl, I Common Activating Groups for NAS Step [1] Addition of the nucleophile (:Nu–) to form a carbanion Addition of the nucleophile (:Nu–) forms a resonance-stabilized carbanion with a new C – Nu bond— three resonance structures can be drawn. • Step is rate-determining • Aromaticity of the benzene ring is lost Step [2] loss of the leaving group re-forms the aromatic ring. • This step is fast because the aromaticity of the benzene ring is restored. ? Explain why a methoxy group (CH3O) increases the rate of electrophilic aromatic substitution, but decreases the rate of nucleophilic aromatic substitution. 2.ArSN1 Mechanism- elimination /addition • This mechanism operates in the reaction of diazonium salts with nucleophiles. •The driving force resides in the strength of the bonding in the nitrogen molecule that makes it a particularly good leaving group. 3.Benzyne Mechanism- elimination /addition Step [1] Elimination of HX to form benzyne Elimination of H and X from two adjacent carbons forms a reactive benzyne intermediate Step [2] Nucleophilic addition to form the substitution product Addition of the nucleophile (–OH in this case) and protonation form the substitution product Evidence for the Benzyne Mechanism Trapping in Diels/Alder Reaction O O B E N Z Y N E C C O O O D i e l s / A l d e r O N H 3 N N Dienophile Diene A d d u c t Substrate Modification – absence of a hydrogens LG Substituent Substituent No Reaction Base Isotopic Labeling LG Nu H Nu Structure of Benzyne • The σ bond is formed by overlap of two sp2 hybrid orbitals.