Autoimmune Hemolytic Anemia in the Pediatric Setting
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Hemolysis and Venous Thrombosis: Which Link? A
ISSN: 2378-3656 Rkiouak et al. Clin Med Rev Case Rep 2020, 7:329 DOI: 10.23937/2378-3656/1410329 Volume 7 | Issue 11 Clinical Medical Reviews Open Access and Case Reports ORIGINAL RESEARCH Hemolysis and Venous Thrombosis: Which Link? A. Rkiouak, PH.D1, I El Kassimi, MD2, N Sahel, MD2, M Zaizaa, MD2 and Y Sekkach, PhD1 Check for updates Internal Medicine A Department, Mohammed V Military Hospital Medical School of Rabat, Morocco *Corresponding author: Adil Rkiouak, PH.D., Department of Internal Medicine A, Mohammed V Military Hospital, Medical School of Rabat, Morocco, Tel: +212-66-179-44-04 The mechanism of antibody-mediated hemolysis is Abstract via phagocytosis or complement-mediated destruction The association hemolysis and venous thrombosis remains and can occur intravascular or extravascular. The intra- unknown to clinicians, despite our advances in comrehen- sion of pathophysiological bases. vascular mechanisms include direct cellular destruction via lysis, toxins, or trauma; fragmentation and oxida- Haemolysis, which is observed in multiple diseases, can affect all three components of Virchow’s triad. It is not sur- tion. prising that there is a link between haemolytic disorders and Multiple haemolytic disorders produce substantial thrombosis. intravascular haemolysis. Examples, the corpuscular We will try to clarify the main pro-thrombotic mechanisms hemolysis include PNH, extra-corpuscular haemolysis, during hemolysis through 3 clinical observations of deep ve- acquired (autoimmune haemolytic anaemia (AIHA , nous thrombosis in 3 main types of hemolytic pathologies, ) namely a case of paroxysmal nocturnal hemoglobinuria, thrombotic thrombocytopenic purpura (PTT)), as well thrombotic thrombocytopenic purpura and autoimmune as other diseases. These disorders are also associated anemia hemolytic. -

Clostridial Septicemia with Intravascular Hemolysis: a Case Report G
Henry Ford Hospital Medical Journal Volume 13 | Number 4 Article 4 12-1965 Clostridial Septicemia With Intravascular Hemolysis: A Case Report G. M. Mastio E. Morfin Follow this and additional works at: https://scholarlycommons.henryford.com/hfhmedjournal Part of the Life Sciences Commons, Medical Specialties Commons, and the Public Health Commons Recommended Citation Mastio, G. M. and Morfin, E. (1965) "Clostridial Septicemia With Intravascular Hemolysis: A Case Report," Henry Ford Hospital Medical Bulletin : Vol. 13 : No. 4 , 421-425. Available at: https://scholarlycommons.henryford.com/hfhmedjournal/vol13/iss4/4 This Article is brought to you for free and open access by Henry Ford Health System Scholarly Commons. It has been accepted for inclusion in Henry Ford Hospital Medical Journal by an authorized editor of Henry Ford Health System Scholarly Commons. Henry Ford Hosp. Med. Bull. Vol. 13, December 1965 CLOSTRIDIAL SEPTICEMIA WITH INTRAVASCULAR HEMOLYSIS A CASE REPORT G. M. MASTIC, M.D. AND E. MORFIN, M.D. In 1871 Bottini' demonstrated the bacterial nature of gas gangrene, but failed to isolate a causal organism. Clostridium perfringens, sometimes known as Clostridium welchii, was discovered independently during 1892 and 1893 by Welch, Frankel, "Veillon and Zuber.^ This organism is a saprophytic inhabitant of the intestinal tract, and may be a harmless saprophyte of the female genital tract occurring in the vagina in 4-6 per cent of pregnant women. Clostridial organisms occur in great numbers and distribution throughout the world. Because of this, they are very common in traumatic wounds. Very few species of Clostridia, however, are pathogenic, and still fewer are capable of producing gas gangrene in man. -

Transfusion Problems in Hemolytic Anemias*
Transfusion Problems in Hemolytic Anemias* ALI A. HOSSAIN! Department of Pathdlogy, Medical College of Virginia, Richmond 23219 All hemolytic anemias feature shortened red cell Extrinsic Mechanisms survival due to premature hemolysis of the cell. For the Those hemolytic anemias which are due to extrinsic purposes of this presentation, we may classify the factors may be classified, further, as non-immune or hemolytic anemias, most broadly, according to the immune. Non-immune mechanisms include a) drugs mechanisms leading to hemolysis. and chemicals (phenylhydrazine, naphthalene, lead, snake venoms); b) physical agents (heat); c) bacteria Intrinsic Mechanisms and parasites (hemolytic streptococci, Clostridium Hemolytic anemias due to intrinsically defective welchii, Bartonella, plasmodia); and d) acquired sen erythrocytes are essentially of three types. First are sitivity to penicillin, methylodopa, Keftin®, or fava those anemias in which the red cells are defective due plant as examples. Some of the agents in this. last to lack of an essential factor, eg, pernicious anemia group serve to lyse the cells, either through duect in relapse. The second type includes those in which action or by formation of antibodies. the red cells have an abnormal shape because of an These hemolytic anemias due to extrinsic factors inherited error in the chemical makeup of the hemo of the non-immune variety present no transfusion globin molecules; eg, sickle cells, elliptocytes, sphero problem for the Blood Bank. However, it ~~st b.e cytes, and the target -
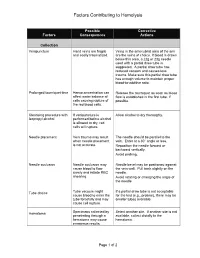
Factors Contributing to Hemolysis
Factors Contributing to Hemolysis Possible Corrective Factors Consequences Actions Collection Venipuncture Hand veins are fragile Veins in the antecubital area of the arm and easily traumatized. are the veins of choice. If blood is drawn below this area, a 22g or 23g needle used with a partial draw tube is suggested. A partial draw tube has reduced vacuum and causes less trauma. Make sure this partial draw tube has enough volume to maintain proper blood-to-additive ratio. Prolonged tourniquet time Hemoconcentration can Release the tourniquet as soon as blood affect water balance of flow is established in the first tube, if cells causing rupture of possible. the red blood cells. Cleansing procedure with If venipuncture is Allow alcohol to dry thoroughly. isopropyl alcohol performed before alcohol is allowed to dry, red cells will rupture. Needle placement Vein trauma may result The needle should be parallel to the when needle placement vein. Enter at a 30° angle or less. is not accurate. Reposition the needle forward or backward vertically. Avoid probing. Needle occlusion Needle occlusion may Needle bevel may be positioned against cause blood to flow the vein wall. Pull back slightly on the slowly and initiate RBC needle. shearing. Avoid rotating or changing the angle of the needle. Tube choice Tube vacuum might If a partial draw tube is not acceptable cause blood to enter the for the test (e.g., protime), there may be tube forcefully and may smaller tubes available. cause cell rupture. Hematoma Specimens collected by Select another site. If another site is not penetrating through a available, collect distally to the hematoma may cause hematoma. -

Predictors of Autoimmune Hemolytic Anemia in Beta-Thalassemia
Blood Cells, Molecules and Diseases 79 (2019) 102342 Contents lists available at ScienceDirect Blood Cells, Molecules and Diseases journal homepage: www.elsevier.com/locate/bcmd Predictors of autoimmune hemolytic anemia in beta-thalassemia patients with underlying red blood cells autoantibodies T ⁎ Monia Ben Khaleda,b, , Monia Ouedernia,b, Nessrine Sahlia,b, Nawel Dhouibb, Ahmed Ben Abdelazizc, Samia Rekayaa,b, Ridha Koukia,b, Houda Kaabid, Hmida Slamad, Fethi Melloulia,b, Mohamed Bejaouia,b a Faculty of Medicine, University of Tunis El Manar, Tunis, Tunisia b Pediatric Immuno-Hematology Unit, Bone Marrow Transplantation Center Tunis, Tunis, Tunisia c Information System Directions, Sahloul University Hospital, Sousse, Tunisia d National Center of Blood Transfusion, Tunis, Tunisia ARTICLE INFO ABSTRACT Editor: Mohandas Narla In beta-thalassemia patients, erythrocyte autoantibodies can remain silent or lead to Autoimmune Hemolytic Keywords: Anemia (AIHA).The aim of this study was to identify predictors of AIHA in beta-thalassemia patients with Autoimmune hemolytic anemia positive Direct Antiglobulin Test (DAT), in Tunisia. Thalassemia This longitudinal prognosis study was carried out on beta-thalassemia patients with a positive confirmed Transfusion DAT. Predictors of AIHA were identified the Kaplan-Meier method. A Cox model analysis was used to identify Autoantibodies independent predictors. Red blood cells Among 385 beta thalassemia patients, 87 developed positive DAT (22.6%). Autoimmune hemolytic anemia Direct antiglobulin test was occurred in 25 patients. Multivariate analysis showed that AIHA was independently associated with beta- thalassemia intermedia and similar family history of AIHA. Splenectomy in patients with positive DAT was independently associated with an increased risk of AIHA (HR = 6.175, CI: 2.049–18.612, p < 0.001). -

Hemolytic Disease of the Newborn
Intensive Care Nursery House Staff Manual Hemolytic Disease of the Newborn INTRODUCTION and DEFINITION: Hemolytic Disease of the Newborn (HDN), also known as erythroblastosis fetalis, isoimmunization, or blood group incompatibility, occurs when fetal red blood cells (RBCs), which possess an antigen that the mother lacks, cross the placenta into the maternal circulation, where they stimulate antibody production. The antibodies return to the fetal circulation and result in RBC destruction. DIFFERENTIAL DIAGNOSIS of hemolytic anemia in a newborn infant: -Isoimmunization -RBC enzyme disorders (e.g., G6PD, pyruvate kinase deficiency) -Hemoglobin synthesis disorders (e.g., alpha-thalassemias) -RBC membrane abnormalities (e.g., hereditary spherocytosis, elliptocytosis) -Hemangiomas (Kasabach Merritt syndrome) -Acquired conditions, such as sepsis, infections with TORCH or Parvovirus B19 (anemia due to RBC aplasia) and hemolysis secondary to drugs. ISOIMMUNIZATION A. Rh disease (Rh = Rhesus factor) (1) Genetics: Rh positive (+) denotes presence of D antigen. The number of antigenic sites on RBCs varies with genotype. Prevalence of genotype varies with the population. Rh negative (d/d) individuals comprise 15% of Caucasians, 5.5% of African Americans, and <1% of Asians. A sensitized Rh negative mother produces anti-Rh IgG antibodies that cross the placenta. Risk factors for antibody production include 2nd (or later) pregnancies*, maternal toxemia, paternal zygosity (D/D rather than D/d), feto-maternal compatibility in ABO system and antigen load. (2) Clinical presentation of HDN varies from mild jaundice and anemia to hydrops fetalis (with ascites, pleural and pericardial effusions). Because the placenta clears bilirubin, the chief risk to the fetus is anemia. Extramedullary hematopoiesis (due to anemia) results in hepatosplenomegaly. -

A Newly Recognized Blood Group in Domestic Shorthair Cats: the Mik Red Cell Antigen
J Vet Intern Med 2007;21:287–292 A Newly Recognized Blood Group in Domestic Shorthair Cats: The Mik Red Cell Antigen Nicole M. Weinstein, Marie-Claude Blais, Kimberly Harris, Donna A. Oakley, Lillian R. Aronson, and Urs Giger Background: Naturally occurring alloantibodies produced against A and B red cell antigens in cats can cause acute hemolytic transfusion reactions. Blood incompatibilities, unrelated to the AB blood group system, have also been suspected after blood transfusions through routine crossmatch testing or as a result of hemolytic transfusion reactions. Hypothesis: Incompatible crossmatch results among AB compatible cats signify the presence of a naturally occurring alloantibody against a newly identified blood antigen in a group of previously never transfused blood donor cats. The associated alloantibody is clinically important based upon a hemolytic transfusion reaction after inadvertent transfusion of red cells expressing this red cell antigen in a feline renal transplant recipient that lacks this red cell antigen. Methods: Blood donor and nonblood donor cats were evaluated for the presence of auto- and alloantibodies using direct antiglobulin and crossmatch tests, respectively, and were blood typed for AB blood group status. Both standard tube and novel gel column techniques were used. Results: Plasma from 3 of 65 cats and 1 feline renal transplant recipient caused incompatible crossmatch test results with AB compatible erythrocytes indicating these cats formed an alloantibody against a red cell antigen they lack, termed Mik. The 3 donors and the renal transplant recipient were crossmatch-compatible with one another. Tube and gel column crossmatch test results were similar. Conclusions and Clinical Importance: The absence of this novel Mik red cell antigen can be associated with naturally occurring anti-Mik alloantibodies and can elicit an acute hemolytic transfusion reaction after an AB-matched blood transfusion. -

Simultaneous Pure Red Cell Aplasia and Auto-Immune Hemolytic Anemia in a Previously Healthy Infant
Simultaneous Pure Red Cell Aplasia and Auto-immune Hemolytic Anemia in a Previously Healthy Infant Robert P. Sanders, MD July 16, 2002 Case Presentation Patient Z.H. • Previously Healthy 7 month old WM • Presented to local ER 6/30 with 1 wk of decreased activity and appetite, low grade temp, 2 day h/o pallor. • Noted to have severe anemia, transferred to LeBonheur • Review of Systems – ? Single episode of dark urine – 4 yo sister diagnosed with Fifth disease 1 wk prior to onset of symptoms, cousin later also diagnosed with Fifth disease – Otherwise negative ROS •PMH – Term, no complications – Normal Newborn Screen – Hospitalized 12/01 with RSV • Medications - None • Allergies - NKDA • FH - Both parents have Hepatitis C (pt negative) • SH - Lives with Mom, 4 yo sister • Development Normal Physical Exam • 37.2 167 33 84/19 9.3kg • Gen - Alert, pale, sl yellow skin tone, NAD •HEENT -No scleral icterus • CHEST - Clear • CV - RRR, II/VI SEM at LLSB • ABD - Soft, BS+, no HSM • SKIN - No Rash • NEURO - No Focal Deficits Labs •CBC – WBC 20,400 • 58% PMN 37% Lymph 4% Mono 1 % Eo – Hgb 3.4 • MCV 75 MCHC 38.0 MCH 28.4 – Platelets 409,000 • Retic 0.5% • Smear - Sl anisocytosis, Sl hypochromia, Mod microcytes, Sl toxic granulation • G6PD Assay 16.6 U/g Hb (nl 4.6-13.5) • DAT, Broad Spectrum Positive – IgG negative – C3b, C3d weakly positive • Chemistries – Total Bili 2.0 – Uric Acid 4.8 –LDH 949 • Urinalysis Negative, Urobilinogen 0.2 • Blood and Urine cultures negative What is your differential diagnosis? Differential Diagnosis • Transient Erythroblastopenia of Childhood • Diamond-Blackfan syndrome • Underlying red cell disorder with Parvovirus induced Transient Aplastic Crisis • Immunohemolytic anemia with reticulocytopenia Hospital Course • Admitted to ICU for observation, transferred to floor 7/1. -

Aplastic Crisis Caused by Parvovirus B19 in an Adult Patient with Sickle-Cell Disease
Revista da Sociedade Brasileira de Medicina Tropical RELATO DE CASO 33(5):477-481, set-out, 2000. Aplastic crisis caused by parvovirus B19 in an adult patient with sickle-cell disease Crise aplástica por parvovírus B19 em um paciente adulto com doença falciforme Sérgio Setúbal1, Adelmo H.D. Gabriel2, Jussara P. Nascimento3 e Solange A. Oliveira1 Abstract We describe a case of aplastic crisis caused by parvovirus B19 in an adult sickle-cell patient presenting with paleness, tiredness, fainting and dyspnea. The absence of reticulocytes lead to the diagnosis. Anti-B19 IgM and IgG were detected. Reticulocytopenia in patients with hereditary hemolytic anemia suggests B19 infection. Key-words: Human parvovirus B19. Sickle-cell disease. Transient aplastic crisis. Reticulocytopenia. Resumo Descreve-se um caso de crise aplástica devida ao parvovírus B19 num paciente adulto, manifestando-se por palidez, cansaço, lipotímias e dispnéia. A ausência de reticulócitos chamou a atenção para o diagnóstico. Detectaram-se IgM e IgG anti-B19. Reticulocitopenia em pacientes com anemia hemolítica hereditária sugere infecção por B19. Palavras-chaves: Parvovírus B19. Doença falciforme. Crise aplástica transitória. Reticulocitopenia. Parvovirus B19 is the only pathogenic and the virus was labeled serum-parvovirus-like parvovirus in humans. It is a DNA virus that infects particle. Retesting the sera from their panels, which and destroys erythroid cell progenitors. Cossart and were obtained mainly from British adults, Cossart coworkers3 discovered parvovirus B19 fortuitously and coworkers demonstrated that 30% of them in 1974, when they were trying to detect HBsAg had antibodies to the virus. in panels of human sera. Unexpectedly, the serum The virus was identified again two years later numbered 19 in panel B showed an anomalous in two blood donors12, and six years later in two precipitin line in a counter immunoelectrophoresis British soldiers returning from Africa15, all of which (CIE) employing another human immune serum. -

A Suspected Case of Clostridium Perfringens Sepsis With
Uojima et al. Journal of Medical Case Reports (2019) 13:125 https://doi.org/10.1186/s13256-019-2023-x CASE REPORT Open Access A suspected case of Clostridium perfringens sepsis with intravascular hemolysis after transhepatic arterial chemoembolization: a case report Haruki Uojima1,2*, Mie Onoue2, Hisashi Hidaka2, Naohisa Wada2, Yoshiaki Tanaka2, Tomoyoshi Inoue2, Kousuke Kubota2, Takahide Nakazawa2, Akitaka Shibuya2 and Wasaburo Koizumi2 Abstract Introduction: Sepsis due to Clostridium perfringens, one of several clostridial species, is an important cause of massive intravascular hemolysis in patients with underlying malignancies. Chronic liver diseases, immunosuppression, and presence of malignancies were risk factors for Clostridium perfringens sepsis. Therefore, Clostridium perfringens sepsis should always be considered in patients presenting with liver damage after chemo- embolic therapy for hepatocellular carcinoma. This case report focuses on findings characteristic of an intravascular hemolysis due to Clostridium perfringens after transhepatic arterial chemoembolization. Case presentation: An 83-year-old Japanese man presented to our hospital because of a third recurrence of hepatocellular carcinoma. He had nonalcoholic steatohepatitis-related cirrhosis, and underwent radiofrequency ablation and transhepatic arterial chemoembolization therapy for hepatocellular carcinoma of S4/S8 and S2. He had a medical history of pancreatic carcinoma and underwent pylorus-preserving pancreaticoduodenectomy approximately 5 years ago. Because follow-up -

Blood Bank I D
The Osler Institute Blood Bank I D. Joe Chaffin, MD Bonfils Blood Center, Denver, CO The Fun Just Never Ends… A. Blood Bank I • Blood Groups B. Blood Bank II • Blood Donation and Autologous Blood • Pretransfusion Testing C. Blood Bank III • Component Therapy D. Blood Bank IV • Transfusion Complications * Noninfectious (Transfusion Reactions) * Infectious (Transfusion-transmitted Diseases) E. Blood Bank V (not discussed today but available at www.bbguy.org) • Hematopoietic Progenitor Cell Transplantation F. Blood Bank Practical • Management of specific clinical situations • Calculations, Antibody ID and no-pressure sample questions Blood Bank I Blood Groups I. Basic Antigen-Antibody Testing A. Basic Red Cell-Antibody Interactions 1. Agglutination a. Clumping of red cells due to antibody coating b. Main reaction we look for in Blood Banking c. Two stages: 1) Coating of cells (“sensitization”) a) Affected by antibody specificity, electrostatic RBC charge, temperature, amounts of antigen and antibody b) Low Ionic Strength Saline (LISS) decreases repulsive charges between RBCs; tends to enhance cold antibodies and autoantibodies c) Polyethylene glycol (PEG) excludes H2O, tends to enhance warm antibodies and autoantibodies. 2) Formation of bridges a) Lattice structure formed by antibodies and RBCs b) IgG isn’t good at this; one antibody arm must attach to one cell and other arm to the other cell. c) IgM is better because of its pentameric structure. P}Chaffin (12/28/11) Blood Bank I page 1 Pathology Review Course 2. Hemolysis a. Direct lysis of a red cell due to antibody coating b. Uncommon, but equal to agglutination. 1) Requires complement fixation 2) IgM antibodies do this better than IgG. -

The Hematological Complications of Alcoholism
The Hematological Complications of Alcoholism HAROLD S. BALLARD, M.D. Alcohol has numerous adverse effects on the various types of blood cells and their functions. For example, heavy alcohol consumption can cause generalized suppression of blood cell production and the production of structurally abnormal blood cell precursors that cannot mature into functional cells. Alcoholics frequently have defective red blood cells that are destroyed prematurely, possibly resulting in anemia. Alcohol also interferes with the production and function of white blood cells, especially those that defend the body against invading bacteria. Consequently, alcoholics frequently suffer from bacterial infections. Finally, alcohol adversely affects the platelets and other components of the blood-clotting system. Heavy alcohol consumption thus may increase the drinker’s risk of suffering a stroke. KEY WORDS: adverse drug effect; AODE (alcohol and other drug effects); blood function; cell growth and differentiation; erythrocytes; leukocytes; platelets; plasma proteins; bone marrow; anemia; blood coagulation; thrombocytopenia; fibrinolysis; macrophage; monocyte; stroke; bacterial disease; literature review eople who abuse alcohol1 are at both direct and indirect. The direct in the number and function of WBC’s risk for numerous alcohol-related consequences of excessive alcohol increases the drinker’s risk of serious Pmedical complications, includ- consumption include toxic effects on infection, and impaired platelet produc- ing those affecting the blood (i.e., the the bone marrow; the blood cell pre- tion and function interfere with blood cursors; and the mature red blood blood cells as well as proteins present clotting, leading to symptoms ranging in the blood plasma) and the bone cells (RBC’s), white blood cells from a simple nosebleed to bleeding in marrow, where the blood cells are (WBC’s), and platelets.