Hidden Transcriptome Signature in Autism Spectrum Disorder (ASD)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Expanding the Phenotypic Spectrum of PAX6 Mutations: from Congenital Cataracts to Nystagmus
G C A T T A C G G C A T genes Article Expanding the Phenotypic Spectrum of PAX6 Mutations: From Congenital Cataracts to Nystagmus Maria Nieves-Moreno 1,* , Susana Noval 1 , Jesus Peralta 1, María Palomares-Bralo 2 , Angela del Pozo 3 , Sixto Garcia-Miñaur 4, Fernando Santos-Simarro 4 and Elena Vallespin 5 1 Department of Ophthalmology, Hospital Universitario La Paz, 28046 Madrid, Spain; [email protected] (S.N.); [email protected] (J.P.) 2 Department of Molecular Developmental Disorders, Medical and Molecular Genetics Institue (INGEMM) IdiPaz, CIBERER, Hospital Universitario La Paz, 28046 Madrid, Spain; [email protected] 3 Department of Bioinformatics, Medical and Molecular Genetics Institue (INGEMM) IdiPaz, CIBERER, Hospital Universitario La Paz, 28046 Madrid, Spain; [email protected] 4 Department of Clinical Genetics, Medical and Molecular Genetics Institue (INGEMM) IdiPaz, CIBERER, Hospital Universitario La Paz, 28046 Madrid, Spain; [email protected] (S.G.-M.); [email protected] (F.S.-S.) 5 Department of Molecular Ophthalmology, Medical and Molecular Genetics Institue (INGEMM) IdiPaz, CIBERER, Hospital Universitario La Paz, 28046 Madrid, Spain; [email protected] * Correspondence: [email protected] Abstract: Background: Congenital aniridia is a complex ocular disorder, usually associated with severe visual impairment, generally caused by mutations on the PAX6 gene. The clinical phenotype of PAX6 mutations is highly variable, making the genotype–phenotype correlations difficult to establish. Methods: we describe the phenotype of eight patients from seven unrelated families Citation: Nieves-Moreno, M.; Noval, with confirmed mutations in PAX6, and very different clinical manifestations. -

Pax6 During Visual System Development
Hedgehog-dependent E3-ligase Midline1 regulates ubiquitin-mediated proteasomal degradation of Pax6 during visual system development Thorsten Pfirrmanna,1, Enrico Jandta,1, Swantje Ranfta,b, Ashwin Lokapallya, Herbert Neuhausa, Muriel Perronc, and Thomas Hollemanna,2 aInstitute for Physiological Chemistry, University of Halle-Wittenberg, 06114 Halle, Germany; bGynecological Hospital, University Medical Center Mannheim, 68167 Mannheim, Germany; and cParis-Saclay Institute of Neuroscience, CNRS, Univ Paris Sud, Université Paris-Saclay, 91405 Orsay, France Edited by Richard M. Harland, University of California, Berkeley, CA, and approved July 19, 2016 (received for review January 16, 2016) Pax6 is a key transcription factor involved in eye, brain, and pancreas remains unclear how Pax6 protein is removed from the eyestalk development. Although pax6 is expressed in the whole prospective territory on time. Some authors report the regulation of Pax6 retinal field, subsequently its expression becomes restricted to the activity by posttranslational modifications (21–23), and most optic cup by reciprocal transcriptional repression of pax6 and pax2. interestingly, Tuoc et al. showed that in cortical progenitor cells, However, it remains unclear how Pax6 protein is removed from the Pax6 protein is degraded by the proteasome mediated by Trim11 eyestalk territory on time. Here, we report that Mid1, a member of (24). However, the existence of similar mechanisms leading to the RBCC/TRIM E3 ligase family, which was first identified in patients the development of the visual system is not known. with the X-chromosome–linked Opitz BBB/G (OS) syndrome, inter- The data of our present study show that Midline1 (Mid1) acts with Pax6. We found that the forming eyestalk is a major do- serves as one of these links. -

Supplementary Materials
Supplementary Materials + - NUMB E2F2 PCBP2 CDKN1B MTOR AKT3 HOXA9 HNRNPA1 HNRNPA2B1 HNRNPA2B1 HNRNPK HNRNPA3 PCBP2 AICDA FLT3 SLAMF1 BIC CD34 TAL1 SPI1 GATA1 CD48 PIK3CG RUNX1 PIK3CD SLAMF1 CDKN2B CDKN2A CD34 RUNX1 E2F3 KMT2A RUNX1 T MIXL1 +++ +++ ++++ ++++ +++ 0 0 0 0 hematopoietic potential H1 H1 PB7 PB6 PB6 PB6.1 PB6.1 PB12.1 PB12.1 Figure S1. Unsupervised hierarchical clustering of hPSC-derived EBs according to the mRNA expression of hematopoietic lineage genes (microarray analysis). Hematopoietic-competent cells (H1, PB6.1, PB7) were separated from hematopoietic-deficient ones (PB6, PB12.1). In this experiment, all hPSCs were tested in duplicate, except PB7. Genes under-expressed or over-expressed in blood-deficient hPSCs are indicated in blue and red respectively (related to Table S1). 1 C) Mesoderm B) Endoderm + - KDR HAND1 GATA6 MEF2C DKK1 MSX1 GATA4 WNT3A GATA4 COL2A1 HNF1B ZFPM2 A) Ectoderm GATA4 GATA4 GSC GATA4 T ISL1 NCAM1 FOXH1 NCAM1 MESP1 CER1 WNT3A MIXL1 GATA4 PAX6 CDX2 T PAX6 SOX17 HBB NES GATA6 WT1 SOX1 FN1 ACTC1 ZIC1 FOXA2 MYF5 ZIC1 CXCR4 TBX5 PAX6 NCAM1 TBX20 PAX6 KRT18 DDX4 TUBB3 EPCAM TBX5 SOX2 KRT18 NKX2-5 NES AFP COL1A1 +++ +++ 0 0 0 0 ++++ +++ ++++ +++ +++ ++++ +++ ++++ 0 0 0 0 +++ +++ ++++ +++ ++++ 0 0 0 0 hematopoietic potential H1 H1 H1 H1 H1 H1 PB6 PB6 PB7 PB7 PB6 PB6 PB7 PB6 PB6 PB6.1 PB6.1 PB6.1 PB6.1 PB6.1 PB6.1 PB12.1 PB12.1 PB12.1 PB12.1 PB12.1 PB12.1 Figure S2. Unsupervised hierarchical clustering of hPSC-derived EBs according to the mRNA expression of germ layer differentiation genes (microarray analysis) Selected ectoderm (A), endoderm (B) and mesoderm (C) related genes differentially expressed between hematopoietic-competent (H1, PB6.1, PB7) and -deficient cells (PB6, PB12.1) are shown (related to Table S1). -

Drosophila Pax6 Promotes Development of the Entire Eye-Antennal Disc, Thereby Ensuring Proper Adult Head Formation
PAPER Drosophila Pax6 promotes development of the entire COLLOQUIUM eye-antennal disc, thereby ensuring proper adult head formation Jinjin Zhua, Sneha Palliyila, Chen Ranb, and Justin P. Kumara,1 aDepartment of Biology, Indiana University, Bloomington, IN 47405; and bDepartment of Biology, Stanford University, Stanford, CA 94305 Edited by Ellen V. Rothenberg, California Institute of Technology, Pasadena, CA, and accepted by Editorial Board Member Neil H. Shubin February 17, 2017 (received for review July 26, 2016) Paired box 6 (Pax6) is considered to be the master control gene for molecular battle among GRNs allows for the subdivision of the eye development in all seeing animals studied so far. In vertebrates, eye-antennal disc to be maintained within a single continuous it is required not only for lens/retina formation but also for the cellular field (13–16). Of the GRNs that are known to operate development of the CNS, olfactory system, and pancreas. Although within the eye-antennal disc, the retinal determination (RD) Pax6 plays important roles in cell differentiation, proliferation, and network, which controls eye development, is the best studied (17). patterning during the development of these systems, the underlying At the core of the RD network lie the Paired box 6 (Pax6) genes mechanism remains poorly understood. In the fruit fly, Drosophila eyeless (ey)andtwin of eyeless (toy), the SIX family member sine melanogaster, Pax6 also functions in a range of tissues, including oculis (so), the transcriptional coactivator eyes absent (eya), and the the eye and brain. In this report, we describe the function of Pax6 in Ski/Sno family member dachshund (dac)(17). -

Distinct Activities of Gli1 and Gli2 in the Absence of Ift88 and the Primary Cilia
Journal of Developmental Biology Article Distinct Activities of Gli1 and Gli2 in the Absence of Ift88 and the Primary Cilia Yuan Wang 1,2,†, Huiqing Zeng 1,† and Aimin Liu 1,* 1 Department of Biology, Eberly College of Sciences, Center for Cellular Dynamics, Huck Institute of Life Science, The Penn State University, University Park, PA 16802, USA; [email protected] (Y.W.); [email protected] (H.Z.) 2 Department of Occupational Health, School of Public Health, China Medical University, No.77 Puhe Road, Shenyang North New Area, Shenyang 110122, China * Correspondence: [email protected]; Tel.: +1-814-865-7043 † These authors contributed equally to this work. Received: 2 November 2018; Accepted: 16 February 2019; Published: 19 February 2019 Abstract: The primary cilia play essential roles in Hh-dependent Gli2 activation and Gli3 proteolytic processing in mammals. However, the roles of the cilia in Gli1 activation remain unresolved due to the loss of Gli1 transcription in cilia mutant embryos, and the inability to address this question by overexpression in cultured cells. Here, we address the roles of the cilia in Gli1 activation by expressing Gli1 from the Gli2 locus in mouse embryos. We find that the maximal activation of Gli1 depends on the cilia, but partial activation of Gli1 by Smo-mediated Hh signaling exists in the absence of the cilia. Combined with reduced Gli3 repressors, this partial activation of Gli1 leads to dorsal expansion of V3 interneuron and motor neuron domains in the absence of the cilia. Moreover, expressing Gli1 from the Gli2 locus in the presence of reduced Sufu has no recognizable impact on neural tube patterning, suggesting an imbalance between the dosages of Gli and Sufu does not explain the extra Gli1 activity. -

Mutational Analysis of PAX6: 16 Novel Mutations Including 5 Missense Mutations with a Mild Aniridia Phenotype
European Journal of Human Genetics (1999) 7, 274–286 t © 1999 Stockton Press All rights reserved 1018–4813/99 $12.00 http://www.stockton-press.co.uk/ejhg ARTICLE Mutational analysis of PAX6: 16 novel mutations including 5 missense mutations with a mild aniridia phenotype Karen Grønskov1, Thomas Rosenberg2, Annie Sand1 and Karen Brøndum-Nielsen1 1Department of Medical Genetics, John F Kennedy Institute, Glostrup 2National Eye Clinic for the Visually Impaired, Hellerup, Denmark Mutations in the developmental control gene PAX6 have been shown to be the genetic cause of aniridia, which is a severe panocular eye disease characterised by iris hypoplasia. The inheritance is autosomal dominant with high penetrance but variable expressivity. Here we describe a mutational analysis of 27 Danish patients using a dideoxy fingerprinting method, which identified PAX6 mutations in 18 individuals with aniridia. A thorough phenotype description was made for the 18 patients. A total of 19 mutations, of which 16 were novel, are described. Among these were five missense mutations which tended to be associated with a milder aniridia phenotype, and in fact one of them seemed to be non-penetrant. Four of the five missense mutations were located in the paired domain. We also describe a third alternative spliced PAX6 isoform in which two of the four missense mutations would be spliced out. Our observations support the concept of dosage effects of PAX6 mutations as well as presenting evidence for variable expressivity and gonadal mosaicism. Keywords: PAX6; aniridia; phenotype–genotype correlation; missense mutations; alternative splicing Introduction but variable expressivity.1 The sporadic cases can be part of the WAGR syndrome (Wilms’ tumour, aniridia, Aniridia is a severe panocular disorder, with a reported genitourinary abnormalities and mental retardation) incidence of 1 in 50 000 to 100 000. -
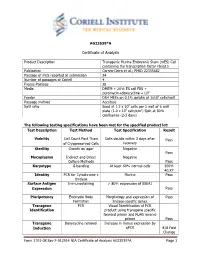
Certificate of Analysis
AG23539*A Certificate of Analysis Product Description Transgenic Murine Embryonic Stem (mES) Cell containing the transcription factor Fbxo15 Publication Correa-Cerro et al.; PMID 22355682 Passage of mES reported at submission 24 Number of passages at Coriell 4 Freeze Passage 28 Media DMEM + 20% ES cell FBS + puromycin+doxycycline + LIF Feeder DR4 MEFs on 0.1% gelatin at 1x105 cells/well Passage method Accutase Split ratio Seed at 1.2 x 106 cells per 1 well of 6 well plate (1.0 x 105 cells/cm2) Split at 80% confluence (2-3 days) The following testing specifications have been met for the specified product lot: Test Description Test Method Test Specification Result Viability Cell Count Post Thaw Cells double within 3 days after Pass of Cryopreserved Cells recovery Sterility Growth on agar Negative Pass Mycoplasma Indirect and Direct Negative Culture Methods Pass Karyotype G-banding At least 60% normal cells 100% 40,XY Identity PCR for Cytochrome c Murine Pass Oxidase Surface Antigen Immunostaining > 80% expression of SSEA1 Expression Pass Pluripotency Embryoid Body Morphology and expression of Pass Formation lineage-specific genes Transgene PCR Visual Identification of PCR Identification product using transgene specific forward primer and FLAG reverse primer Pass Transgene Doxycycline removal Increase in Venus expression by Induction qPCR 418 Fold Change Form 1701-08 Rev F-012914 NIA Certificate of Analysis AG23539*A Page 1 Post-Thaw Viability One vial was thawed after cryopreservation. Cells are counted following recovery and plated in one well of a 6 well plate. Cultures are observed daily and passaged when cells are approximately 80% confluent. -

PDF Download
Review Xatzipsalti Maria et al. Congenital Hypopituitarism: Various Genes, … Horm Metab Res 2018; 00: 00–00 Congenital Hypopituitarism: Various Genes, Various Phenotypes Authors Maria Xatzipsalti1, 2, Antonis Voutetakis1, Lela Stamoyannou2, George P. Chrousos1, Christina Kanaka-Gantenbein1 Affiliations ABSTRacT 1 Division of Endocrinology, Diabetes and Metabolism, The ontogenesis and development of the pituitary gland is a First Department of Pediatrics, Medical School, National highly complex process that depends on a cascade of transcrip- and Kapodistrian University of Athens, “Aghia Sofia” tion factors and signaling molecules. Spontaneous mutations Children's Hospital, Athens, Greece and transgenic murine models have demonstrated a role for 2 First Department of Pediatrics, “Aglaia Kyriakou” many of these factors, including HESX1, PROP1, PIT1, LHX3, Children's Hospital, Athens, Greece LHX4, SOX2, SOX3, OTX2, PAX6, FGFR1, SHH, GLI2, and FGF8 in the etiology of congenital hypopituitarism. Genetic muta- Key words tions in any of these factors can lead to congenital hypopitui- pituitary, combined pituitary hormone deficiency, congenital tarism, which is characterized by the deficiency in one or more hypopituitarism, transcription factors, syndromic hypopitui- pituitary hormones. The phenotype can be highly variable, tarism, non-syndromic hypopituitarism consisting of isolated hypopituitarism or more complex disor- ders. The same phenotype can be attributed to different gene received 27.03.2018 mutations; while a given gene mutation can -

The Identification and Characterization of Estrogen
Duquesne University Duquesne Scholarship Collection Electronic Theses and Dissertations Fall 2009 The deI ntification and Characterization of Estrogen Receptors in the Mouse and Human Lens and Their Role in Cataract Development Mary Rachel Kirker Follow this and additional works at: https://dsc.duq.edu/etd Recommended Citation Kirker, M. (2009). The deI ntification and Characterization of Estrogen Receptors in the Mouse and Human Lens and Their Role in Cataract Development (Doctoral dissertation, Duquesne University). Retrieved from https://dsc.duq.edu/etd/750 This Immediate Access is brought to you for free and open access by Duquesne Scholarship Collection. It has been accepted for inclusion in Electronic Theses and Dissertations by an authorized administrator of Duquesne Scholarship Collection. For more information, please contact [email protected]. THE IDENTIFICATION AND CHARACTERIZATION OF ESTROGEN RECEPTORS IN THE MOUSE AND HUMAN LENS AND THEIR ROLE IN CATARACT DEVELOPMENT A Dissertation Submitted to the Graduate School of Pharmaceutical Sciences Duquesne University In partial fulfillment of the requirements for the degree of Doctor of Philosophy By M. Rachel Kirker December 2009 Copyright by M. Rachel Kirker 2009 THE IDENTIFICATION AND CHARACTERIZATION OF ESTROGEN RECEPTORS IN THE MOUSE AND HUMAN LENS AND THEIR ROLE IN CATARACT DEVELOPMENT By M. Rachel Kirker Approved March 27, 2009 ________________________________ ________________________________ Vicki L. Davis, Ph.D. Jane E. Cavanaugh, Ph.D. Dissertation Chair Committee Member Assistant Professor of Pharmacology Assistant Professor of Pharmacology Graduate School of Pharmaceutical Sciences Graduate School of Pharmaceutical Science Duquesne University Duquesne University Pittsburgh, Pennsylvania Pittsburgh, Pennsylvania ________________________________ ________________________________ Carmen M.H. Colitz, D.V.M., Ph.D. -

Two Independent and Interactive DNA-Binding Subdomains of the Pax6 Paired Domain Are Regulated by Alternative Splicing
Downloaded from genesdev.cshlp.org on October 8, 2021 - Published by Cold Spring Harbor Laboratory Press Two independent and interactive DNA-binding subdomains of the Pax6 paired domain are regulated by alternative splicing Jonathan A. Epstein, 1'2 Tom Glaser, 2 Jiexing Cai, 2 Lisa Jepeal, 2 David S. Walton, 3 and Richard L. Maas 2"4 Divisions of Cardiology~ and Genetics2, Department of Medicine and Howard Hughes Medical Institute, Brigham and Women's Hospital, and Massachusetts Eye and Ear Infirmary, a Harvard Medical School, Boston, Massachusetts 02115 USA Vertebrate Pax proteins share a conserved 128-amino-acid DNA-binding motif, the paired domain. The PAX6 gene, which is mutated in the routine Small eye and human aniridia developmental defects, also encodes a second protein with a 14-amino-acid insertion in the paired domain. This protein, which arises by alternative mRNA splicing, exhibits unique DNA-binding properties. Unlike other paired domains, which bind DNA predominantly by their amino termini, the extended Pax6 paired domain interacts with DNA exclusively through its carboxyl terminus. This property can be simulated by deletion of 30 amino-terminal residues from the Pax6 or Pax2 paired domains. Thus, the insertion acts as a molecular toggle to unmask the DNA-binding potential of the carboxyl terminus. The functional nonequivalence of the two Pax6 proteins is underscored by a T ~ C mutation at position -3 of the alternative splice acceptor site that changes the ratio of the two isoforms and causes a distinct human ocular syndrome. [Key Words: Pax genes; transcription factor; alternative mRNA splicing; DNA binding; ocular development; aniridia] Received May 17, 1994; revised version accepted July 18, 1994. -

In Vivo and in Vitro Analysis of Dll1 and Pax6 Function in the Adult Mouse Pancreas
TECHNISCHE UNIVERSITÄT MÜNCHEN Lehrstuhl für Experimentelle Genetik In vivo and in vitro analysis of Dll1 and Pax6 function in the adult mouse pancreas Davide Cavanna Vollständiger Abdruck der von der Fakultät Wissenschaftszentrum Weihenstephan für Ernährung, Landnutzung und Umwelt der Technischen Universität München zur Erlangung des akademischen Grades eines Doktors der Naturwissenschaften genehmigten Dissertation. Vorsitzender: Univ.-Prof. Dr. D. Langosch Prüfer der Dissertation: 1. Univ.-Prof. Dr. M. Hrabé de Angelis 2. Univ.-Prof. A. Schnieke, Ph.D. Die Dissertation wurde am 03.07.2013 bei der Technischen Universität München eingereicht und durch die Fakultät Wissenschaftszentrum Weihenstephan für Ernährung, Landnutzung und Umwelt am 10.12.2013 angenommen. I. Table of contents I. TABLE OF CONTENTS .................................................................................................. I II. FIGURES AND TABLES ................................................................................................ V III. ABBREVIATIONS ................................................................................................. VIII IV. PUBLICATIONS, TALKS, AND POSTERS ................................................................... XI V. ACKNOWLEDGMENTS .............................................................................................. XII VI. AFFIRMATION ..................................................................................................... XIV 1. SUMMARY/ZUSAMMENFASSUNG ............................................................................ -

Blocking Signaling at the Level of GLI Regulates Downstream Gene Expression and Inhibits Proliferation of Canine Osteosarcoma Cells
Blocking Signaling at the Level of GLI Regulates Downstream Gene Expression and Inhibits Proliferation of Canine Osteosarcoma Cells Mehdi Hayat Shahi, Roseline Holt, Robert B. Rebhun* The Department of Surgical and Radiological Sciences, University of California Davis School of Veterinary Medicine, Davis, California, United States of America Abstract The Hedgehog-GLI signaling pathway is active in a variety of human malignancies and is known to contribute to the growth and survival of human osteosarcoma cells. In this study, we examined the expression and regulation of GLI transcription factors in multiple canine osteosarcoma cell lines and analyzed the effects of inhibiting GLI with GANT61, a GLI-specific inhibitor. Compared with normal canine osteoblasts, real-time PCR showed that GLI1 and GLI2 were highly expressed in two out of three cell lines and correlated with downstream target gene expression of PTCH1and PAX6. Treatment of canine osteosarcoma cells with GANT61 resulted in decreased expression of GLI1, GLI2, PTCH1, and PAX6. Furthermore, GANT61 inhibited proliferation and colony formation in all three canine osteosarcoma cell lines. The finding that GLI signaling activity is present and active in canine osteosarcoma cells suggests that spontaneously arising osteosarcoma in dogs might serve as a good model for future preclinical testing of GLI inhibitors. Citation: Shahi MH, Holt R, Rebhun RB (2014) Blocking Signaling at the Level of GLI Regulates Downstream Gene Expression and Inhibits Proliferation of Canine Osteosarcoma Cells. PLoS ONE 9(5): e96593. doi:10.1371/journal.pone.0096593 Editor: Joseph Alan Bauer, Bauer Research Foundation, United States of America Received August 8, 2013; Accepted April 9, 2014; Published May 8, 2014 Copyright: ß 2014 Shahi et al.