Expanding the Phenotypic Spectrum of PAX6 Mutations: from Congenital Cataracts to Nystagmus
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Treacher Collins Prize Essay the Significance of Nystagmus
Eye (1989) 3, 816--832 Treacher Collins Prize Essay The Significance of Nystagmus NICHOLAS EVANS Norwich Introduction combined. The range of forms it takes, and Ophthalmology found the term v!to"[<xy!too, the circumstances in which it occurs, must be like many others, in classical Greece, where it compared and contrasted in order to under described the head-nodding of the wined and stand the relationships between nystagmus of somnolent. It first acquired a neuro-ophthal different aetiologies. An approach which is mological sense in 1822, when it was used by synthetic as well as analytic identifies those Goodl to describe 'habitual squinting'. Since features which are common to different types then its meaning has been refined, and much and those that are distinctive, and helps has been learned about the circumstances in describe the relationship between eye move which the eye oscillates, the components of ment and vision in nystagmus. nystagmus, and its neurophysiological, Nystagmus is not properly a disorder of eye neuroanatomic and neuropathological corre movement, but one of steady fixation, in lates. It occurs physiologically and pathologi which the relationship between eye and field cally, alone or in conjunction with visual or is unstable. The essential significance of all central nervous system pathology. It takes a types of nystagmus is the disturbance in this variety of different forms, the eyes moving relationship between the sensory and motor about one or more axis, and may be conjugate ends of the visual-oculomotor axis. Optimal or dysjugate. It can be modified to a variable visual performance requires stability of the degree by external (visual, gravitational and image on the retina, and vision is inevitably rotational) and internal (level of awareness affected by nystagmus. -

Congenital Cataract
3801 W. 15th St., Bldg. A, Ste. 110 Plano, TX 75075 Phone: (972) 758-0625 Fax: (972) 964-5725 Email: [email protected] Website: www.drstagerjr.com Congenital Cataract Your eye works a lot like a camera. Light rays focus through the lens on the retina, a layer of light-sensitive cells at the back of the eye. Similar to photographic film, the retina allows the image to be “seen” by the brain. Over time, the lens of our eye can become cloudy, preventing light rays from passing clearly through the lens. The loss of transparency may be so mild that vision is barely affected, or it can be so severe that no shapes or movements are seen—only light and dark. When the lens becomes cloudy enough to obstruct vision to any significant degree, it is called a cataract. Eyeglasses or contact lenses can usually correct slight refractive errors caused by early cataracts, but they cannot sharpen your vision if a severe cataract is present. The most common cause of cataract is aging. Occasionally, babies are born with cataracts or develop them very early in life. This condition is called congenital cataract. There are many causes of congenital cataract. Certain diseases can cause the condition, and sometimes it can be inherited. However, in most cases, there is no identifiable cause. Treatment for cataract in infants varies depending on the nature of each patient’s condition. Surgery is usually recommended very early in life, but many factors affect this decision, including the infant’s health and whether there is a cataract in one or both eyes. -

In Vivo Studies Using the Classical Mouse Diversity Panel
The Mouse Diversity Panel Predicts Clinical Drug Toxicity Risk Where Classical Models Fail Alison Harrill, Ph.D The Hamner-UNC Institute for Drug Safety Sciences 0 The Importance of Predicting Clinical Adverse Drug Reactions (ADR) Figure: Cath O’Driscoll Nature Publishing 2004 Risk ID PGx Testing 1 People Respond Differently to Drugs Pharmacogenetic Markers Identified by Genome-Wide Association Drug Adverse Drug Risk Allele Reaction (ADR) Abacavir Hypersensitivity HLA-B*5701 Flucloxacillin Hepatotoxicity Allopurinol Cutaneous ADR HLA-B*5801 Carbamazepine Stevens-Johnson HLA-B*1502 Syndrome Augmentin Hepatotoxicity DRB1*1501 Ximelagatran Hepatotoxicity DRB1*0701 Ticlopidine Hepatotoxicity HLA-A*3303 Average preclinical populations and human hepatocytes lack the diversity to detect incidence of adverse events that occur only in 1/10,000 people. Current Rodent Models of Risk Assessment The Challenge “At a time of extraordinary scientific progress, methods have hardly changed in several decades ([FDA] 2004)… Toxicologists face a major challenge in the twenty-first century. They need to embrace the new “omics” techniques and ensure that they are using the most appropriate animals if their discipline is to become a more effective tool in drug development.” -Dr. Michael Festing Quantitative geneticist Toxicol Pathol. 2010;38(5):681-90 Rodent Models as a Strategy for Hazard Characterization and Pharmacogenetics Genetically defined rodent models may provide ability to: 1. Improve preclinical prediction of drugs that carry a human safety risk 2. -

Human Transcription Factor Protein-Protein Interactions in Health and Disease
HELKA GÖÖS GÖÖS HELKA Recent Publications in this Series 45/2019 Mgbeahuruike Eunice Ego Evaluation of the Medicinal Uses and Antimicrobial Activity of Piper guineense (Schumach & Thonn) 46/2019 Suvi Koskinen AND DISEASE IN HEALTH INTERACTIONS PROTEIN-PROTEIN FACTOR HUMAN TRANSCRIPTION Near-Occlusive Atherosclerotic Carotid Artery Disease: Study with Computed Tomography Angiography 47/2019 Flavia Fontana DISSERTATIONES SCHOLAE DOCTORALIS AD SANITATEM INVESTIGANDAM Biohybrid Cloaked Nanovaccines for Cancer Immunotherapy UNIVERSITATIS HELSINKIENSIS 48/2019 Marie Mennesson Kainate Receptor Auxiliary Subunits Neto1 and Neto2 in Anxiety and Fear-Related Behaviors 49/2019 Zehua Liu Porous Silicon-Based On-Demand Nanohybrids for Biomedical Applications 50/2019 Veer Singh Marwah Strategies to Improve Standardization and Robustness of Toxicogenomics Data Analysis HELKA GÖÖS 51/2019 Iryna Hlushchenko Actin Regulation in Dendritic Spines: From Synaptic Plasticity to Animal Behavior and Human HUMAN TRANSCRIPTION FACTOR PROTEIN-PROTEIN Neurodevelopmental Disorders 52/2019 Heini Liimatta INTERACTIONS IN HEALTH AND DISEASE Efectiveness of Preventive Home Visits among Community-Dwelling Older People 53/2019 Helena Karppinen Older People´s Views Related to Their End of Life: Will-to-Live, Wellbeing and Functioning 54/2019 Jenni Laitila Elucidating Nebulin Expression and Function in Health and Disease 55/2019 Katarzyna Ciuba Regulation of Contractile Actin Structures in Non-Muscle Cells 56/2019 Sami Blom Spatial Characterisation of Prostate Cancer by Multiplex -

"Nystagmus Testing in Intoxicated Individuals," Citek
ISSUE HIGHLIGHT Nystagmus testing in intoxicated individuals Karl Citek, O.D., Ph.D.,a Bret Ball, O.D.,a and Dale A. Rutledge, Lieutenantb aCollege of Optometry, Pacific University, Forest Grove, Oregon and bthe Oregon State Police, Wilsonville, Oregon Background: Law enforcement officers routinely conduct psy- n the United States, drivers impaired* by alcohol and/or chophysical tests to determine if an impaired driver may be drugs are responsible for more than 16,000 deaths, one intoxicated or in need of medical assistance. Testing includes million injuries, and $45 billion in costs annually.1 As assessment of eye movements, using the Horizontal Gaze Nys- I tagmus (HGN) and Vertical Gaze Nystagmus (VGN) tests, which part of the attempt to reduce these human and economic are conducted at roadside by patrol officers. These tests pre- tolls, law enforcement officers routinely conduct tests of eye viously have been validated when the subject is placed in a movements to determine if a driver is under the influence standing posture with head upright. However, certain condi- of alcohol or other drugs. Alcohol, other central nervous sys- tions require that the subject be tested while seated or supine. Under these conditions, Positional Alcohol Nystagmus (PAN) tem (CNS)-depressant drugs, inhalants, and phencyclidine could be induced and mistaken for HGN or VGN. (PCP) and its analogs will affect the neural centers in the Methods: The study was conducted at law enforcement train- brainstem and cerebellum, which control eye movements, ing academy alcohol workshops in the Pacific Northwest. as well as other motor, sensory, and cognitive integration Ninety-six volunteer drinkers were tested when sober and areas of the brain. -

Complex Strabismus and Syndromes
Complex Strabismus & Syndromes Some patients exhibit complex combinations of vertical, horizontal, and torsional strabismus. Dr. Shin treats patients with complex strabismus arising from, but not limited to, thyroid-related eye disease, stroke, or brain tumors as well as strabismic disorders following severe orbital and head trauma. The following paragraphs describe specific ocular conditions marked by complex strabismus. Duane Syndrome Duane syndrome represents a constellation of eye findings present at birth that results from an absent 6th cranial nerve nucleus and an aberrant branch of the 3rd cranial nerve that innervates the lateral rectus muscle. Duane syndrome most commonly affects the left eye of otherwise healthy females. Duane syndrome includes several variants of eye movement abnormalities. In the most common variant, Type I, the eye is unable to turn outward to varying degrees from the normal straight ahead position. In addition, when the patient tries to look straight ahead, the eyes may cross. This may lead a person with Duane syndrome to turn his/her head toward one side while viewing objects in front of them in order to better align the eyes. When the involved eye moves toward the nose, the eye retracts slightly back into the eye socket causing a narrowing of the opening between the eyelids. In Type II, the affected eye possesses limited ability to turn inward and is generally outwardly turning. In Type III, the eye has limited inward and outward movement. All three types are characterized by anomalous co-contraction of the medial and lateral rectus muscles, so when the involved eye moves towards the nose, the globe pulls back into the orbit and the vertical space between the eyelids narrows. -

Sureselect Community Design Glasgow Cancer Panels
SureSelect Community Design Glasgow Cancer Panels Biomarkers with proven or emerging utility Genomics is a key element in precision medicine's potential to transform in predicting outcome and/or drug response/resistance - The clinical space oncology. Hybrid capture-based, targeted next-generation sequencing (NGS) represents a particularly promising technology as it enables the focused profiling of each of the many cancer-relevant loci. This approach allows for Ca rapid and cost-effective detection of most cancer-relevant genomic events, nc Cancer Core with the added advantage of FFPE tissue sample compatibility. er The Glasgow Precision Oncology Laboratory (GPOL) is a team of scientists Haem Cancer Plus with internationally recognized expertise in the technology, biology, and clinical utility of cancer genomics. Among their achievements is their detailed curation of genomic data to define the landscape of clinically and biologically significant genomic events in cancer. This includes not only a published literature review (see references 1-3), but also the International Cancer High confidence cancer genes, including those that are not currently clinically Genome Consortium (ICGC) and the Pan-Cancer Analysis of Whole Genomes actionable - The development space (PCAWG) study. Figure 1. Illustration of the GPOL cancer assay suite of panels that are commercially available as part of The GPOL has leveraged these insights to design the hybrid capture-based Agilent's Community Designs. We currently offer the SureSelect Community Design (CD) Cancer Core, Cancer SureSelect cancer NGS panels. With the SureSelect platform, the GPOL team Plus, and Cancer Haem panels. This figure illustrates the has developed a suite of affordable, fit-for-purpose cancer genomic assays relative size and content overlap of each panel. -

Sixth Nerve Palsy
COMPREHENSIVE OPHTHALMOLOGY UPDATE VOLUME 7, NUMBER 5 SEPTEMBER-OCTOBER 2006 CLINICAL PRACTICE Sixth Nerve Palsy THOMAS J. O’DONNELL, MD, AND EDWARD G. BUCKLEY, MD Abstract. The diagnosis and etiologies of sixth cranial nerve palsies are reviewed along with non- surgical and surgical treatment approaches. Surgical options depend on the function of the paretic muscle, the field of greatest symptoms, and the likelihood of inducing diplopia in additional fields by a given procedure. (Comp Ophthalmol Update 7: xx-xx, 2006) Key words. botulinum toxin (Botox®) • etiology • sixth nerve palsy (paresis) Introduction of the cases, the patients had hypertension and/or, less frequently, Sixth cranial nerve (abducens) palsy diabetes; 26% were undetermined, is a common cause of acquired 5% had a neoplasm, and 2% had an horizontal diplopia. Signs pointing aneurysm. It was noted that patients toward the diagnosis are an who had an aneurysm or neoplasm abduction deficit and an esotropia had additional neurologic signs or increasing with gaze toward the side symptoms or were known to have a of the deficit (Figure 1). The diplopia cancer.2 is typically worse at distance. Measurements are made with the Anatomical Considerations uninvolved eye fixing (primary deviation), and will be larger with the The sixth cranial nerve nuclei are involved eye fixing (secondary located in the lower pons beneath the deviation). A small vertical deficit may fourth ventricle. The nerve on each accompany a sixth nerve palsy, but a side exits from the ventral surface of deviation over 4 prism diopters the pons. It passes from the posterior Dr. O’Donnell is affiliated with the should raise the question of cranial fossa to the middle cranial University of Tennessee Health Sci- additional pathology, such as a fourth fossa, ascends the clivus, and passes ence Center, Memphis, TN. -

Prominent and Regressive Brain Developmental Disorders Associated with Nance-Horan Syndrome
brain sciences Article Prominent and Regressive Brain Developmental Disorders Associated with Nance-Horan Syndrome Celeste Casto 1,†, Valeria Dipasquale 1,†, Ida Ceravolo 2, Antonella Gambadauro 1, Emanuela Aliberto 3, Karol Galletta 4, Francesca Granata 4, Giorgia Ceravolo 1, Emanuela Falzia 5, Antonella Riva 6 , Gianluca Piccolo 6, Maria Concetta Cutrupi 1, Pasquale Striano 6,7 , Andrea Accogli 7,8, Federico Zara 7,8, Gabriella Di Rosa 9, Eloisa Gitto 10, Elisa Calì 11, Stephanie Efthymiou 11 , Vincenzo Salpietro 6,7,11,*, Henry Houlden 11 and Roberto Chimenz 12 1 Department of Human Pathology in Adult and Developmental Age “Gaetano Barresi”, Unit of Emergency Pediatric, University of Messina, Via Consolare Valeria 1, 98125 Messina, Italy; [email protected] (C.C.); [email protected] (V.D.); [email protected] (A.G.); [email protected] (G.C.); [email protected] (M.C.C.) 2 Unit of Ophthalmology, Department of Clinical and Experimental Medicine, University of Messina, Via Consolare Valeria 1, 98125 Messina, Italy; [email protected] 3 Casa di Cura la Madonnina, Via Quadronno 29, 20122 Milano, Italy; [email protected] 4 Department of Biomedical, Dental Science and Morphological and Functional Images, Neuroradiology Unit, University of Messina, Via Consolare Valeria 1, 98125 Messina, Italy; [email protected] (K.G.); [email protected] (F.G.) 5 Azienza Ospedaliera di Cosenza, Via San Martino, 87100 Cosenza, Italy; [email protected] 6 Pediatric Neurology and Muscular Diseases Unit, -

Insertion of Aqueous Shunt in Pedicatric Glaucoma
1/29/2018 Challenges of Insertion of Aqueous shunt in paediatric glaucoma Ahmed Elkarmouty MD, FRCS Moorfields Eye Hospital London, UK Classification • Primary Childhood Glaucoma • A- Primary Congenital Glaucoma (PCG) 1: 10,000–18,000 • B- Juvenile Open Angle Glaucoma (JOAG) (5-35 ys,)1 : 50,000. • Secondary Childhood Glaucoma • A- Glaucoma associated with non-acquired ocular anomalies • B- Glaucoma associated with non- acquired systemic disease or syndrome • C- Glaucoma associated with acquired condition • D- Glaucoma following Cataract surgery 1 1/29/2018 Glaucoma associated with non- acquired ocular anomalies • Conditions with predominantly ocular anomalies present at birth which may or may not be associated with systemic signs • Axenfeld Reiger anomaly • Peters anomaly • Ectropion Uvae • Congenital iris hypolplasia • Aniridia • Oculodermal melanocytosis • Posterior polymorphous dystrophy • Microphthalmos • Microcornea • Ectopia Lentis ( et pupillae) • Persistent foetus vasculopathy Glaucoma associated with non- acquired systemic disease or syndrome predominantly associated with known syndrome, systemic anomalies present at birth which may be associated with ocular signs • Down Syndrome • Connective tissue disorder: Marfan syndrome, Weill- Marchesiani syndrome, Stickler syndrome • Metabolic disorder : Homocystenuria, lowe syndrome, Mucoploysacchroidoses • Phacomatoses: Neurofibromatoses, Sturge Weber, Klipple-Trenaunay- weber syndrome, Rubenstein Taybi • Congenital Rubella 2 1/29/2018 Glaucoma associated with acquired condition Conditions -
Solved/Unsolved
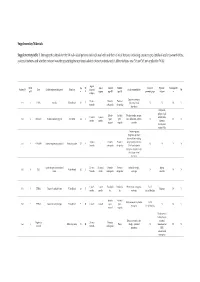
Supplementary Materials: Supplementary table 1. Demographic details for the 54 individual patients (solved/unsolved) and their clinical features including cataract type, details of ocular co-morbidities, systemic features and whether cataract was the presenting feature (non-isolated cataract patients only). Abbreviations: yes (Y), no (N), not applicable (N/A). Age at Famil Ag M/ Age at Cataract Cataract Cataract Systemic Consanguinit Patient ID Gene Confirmed genetic diagnosis Ethnicity diagnosi Ocular co-morbidities FH y ID e F surgery type RE type LE presenting sign features y s (days) Aniridia, nystagmus, 23 years Posterior Posterior 1-1 1 PAX6 Aniridia White British 25 F - glaucoma, foveal N N N Y 4 months subcapsular subcapsular hypoplasia Cleft palate, epilepsy, high Aphakia Aphakia Macular atrophy, myopia, 7 years 9 7 years 8 arched palate, 2-1 2 COL11A1 Stickler syndrome, type II Not Stated 34 F (post- (post- lens subluxation, vitreous N N N months months flattened surgical) surgical) anomaly maxilla, short stature (5'2ft) Anterior segment dysgenesis, pupillary abnormalities including 12 years Posterior Posterior ectopic pupils, ectropion 3-1 3 CPAMD8 Anterior segment dysgenesis 8 Other, Any other 27 F - N N Y N 5 months subcapsular subcapsular UVAE and irodensis, nystagmus, dysplastic optic discs, large corneal diameters Gyrate atrophy of choroid and 23 years 29 years 1 Posterior Posterior Retinal dystrophy, Bipolar 4-1 4 OAT White British 42 F N N N retina 7 months month subcapsular subcapsular exotropia disorder 1 year 6 1 year -

Pax6 During Visual System Development
Hedgehog-dependent E3-ligase Midline1 regulates ubiquitin-mediated proteasomal degradation of Pax6 during visual system development Thorsten Pfirrmanna,1, Enrico Jandta,1, Swantje Ranfta,b, Ashwin Lokapallya, Herbert Neuhausa, Muriel Perronc, and Thomas Hollemanna,2 aInstitute for Physiological Chemistry, University of Halle-Wittenberg, 06114 Halle, Germany; bGynecological Hospital, University Medical Center Mannheim, 68167 Mannheim, Germany; and cParis-Saclay Institute of Neuroscience, CNRS, Univ Paris Sud, Université Paris-Saclay, 91405 Orsay, France Edited by Richard M. Harland, University of California, Berkeley, CA, and approved July 19, 2016 (received for review January 16, 2016) Pax6 is a key transcription factor involved in eye, brain, and pancreas remains unclear how Pax6 protein is removed from the eyestalk development. Although pax6 is expressed in the whole prospective territory on time. Some authors report the regulation of Pax6 retinal field, subsequently its expression becomes restricted to the activity by posttranslational modifications (21–23), and most optic cup by reciprocal transcriptional repression of pax6 and pax2. interestingly, Tuoc et al. showed that in cortical progenitor cells, However, it remains unclear how Pax6 protein is removed from the Pax6 protein is degraded by the proteasome mediated by Trim11 eyestalk territory on time. Here, we report that Mid1, a member of (24). However, the existence of similar mechanisms leading to the RBCC/TRIM E3 ligase family, which was first identified in patients the development of the visual system is not known. with the X-chromosome–linked Opitz BBB/G (OS) syndrome, inter- The data of our present study show that Midline1 (Mid1) acts with Pax6. We found that the forming eyestalk is a major do- serves as one of these links.