Glaucoma and Frequency of Ocular and General Diseases in 30 Patients with Aniridia: a Clinical Study
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Bass – Glaucomatous-Type Field Loss Not Due to Glaucoma
Glaucoma on the Brain! Glaucomatous-Type Yes, we see lots of glaucoma Field Loss Not Due to Not every field that looks like glaucoma is due to glaucoma! Glaucoma If you misdiagnose glaucoma, you could miss other sight-threatening and life-threatening Sherry J. Bass, OD, FAAO disorders SUNY College of Optometry New York, NY Types of Glaucomatous Visual Field Defects Paracentral Defects Nasal Step Defects Arcuate and Bjerrum Defects Altitudinal Defects Peripheral Field Constriction to Tunnel Fields 1 Visual Field Defects in Very Early Glaucoma Paracentral loss Early superior/inferior temporal RNFL and rim loss: short axons Arcuate defects above or below the papillomacular bundle Arcuate field loss in the nasal field close to fixation Superotemporal notch Visual Field Defects in Early Glaucoma Nasal step More widespread RNFL loss and rim loss in the inferior or superior temporal rim tissue : longer axons Loss stops abruptly at the horizontal raphae “Step” pattern 2 Visual Field Defects in Moderate Glaucoma Arcuate scotoma- Bjerrum scotoma Focal notches in the inferior and/or superior rim tissue that reach the edge of the disc Denser field defects Follow an arcuate pattern connected to the blind spot 3 Visual Field Defects in Advanced Glaucoma End-Stage Glaucoma Dense Altitudinal Loss Progressive loss of superior or inferior rim tissue Non-Glaucomatous Etiology of End-Stage Glaucoma Paracentral Field Loss Peripheral constriction Hereditary macular Loss of temporal rim tissue diseases Temporal “islands” Stargardt’s macular due -

Optic Nerve Hypoplasia Plus: a New Way of Looking at Septo-Optic Dysplasia
Optic Nerve Hypoplasia Plus: A New Way of Looking at Septo-Optic Dysplasia Item Type text; Electronic Thesis Authors Mohan, Prithvi Mrinalini Publisher The University of Arizona. Rights Copyright © is held by the author. Digital access to this material is made possible by the University Libraries, University of Arizona. Further transmission, reproduction or presentation (such as public display or performance) of protected items is prohibited except with permission of the author. Download date 29/09/2021 22:50:06 Item License http://rightsstatements.org/vocab/InC/1.0/ Link to Item http://hdl.handle.net/10150/625105 OPTIC NERVE HYPOPLASIA PLUS: A NEW WAY OF LOOKING AT SEPTO-OPTIC DYSPLASIA By PRITHVI MRINALINI MOHAN ____________________ A Thesis Submitted to The Honors College In Partial Fulfillment of the Bachelors degree With Honors in Physiology THE UNIVERSITY OF ARIZONA M A Y 2 0 1 7 Approved by: ____________________________ Dr. Vinodh Narayanan Center for Rare Childhood Disorders Abstract Septo-optic dysplasia (SOD) is a rare congenital disorder that affects 1/10,000 live births. At its core, SOD is a disorder resulting from improper embryological development of mid-line brain structures. To date, there is no comprehensive understanding of the etiology of SOD. Currently, SOD is diagnosed based on the presence of at least two of the following three factors: (i) optic nerve hypoplasia (ii) improper pituitary gland development and endocrine dysfunction and (iii) mid-line brain defects, including agenesis of the septum pellucidum and/or corpus callosum. A literature review of existing research on the disorder was conducted. The medical history and genetic data of 6 patients diagnosed with SOD were reviewed to find damaging variants. -

Insertion of Aqueous Shunt in Pedicatric Glaucoma
1/29/2018 Challenges of Insertion of Aqueous shunt in paediatric glaucoma Ahmed Elkarmouty MD, FRCS Moorfields Eye Hospital London, UK Classification • Primary Childhood Glaucoma • A- Primary Congenital Glaucoma (PCG) 1: 10,000–18,000 • B- Juvenile Open Angle Glaucoma (JOAG) (5-35 ys,)1 : 50,000. • Secondary Childhood Glaucoma • A- Glaucoma associated with non-acquired ocular anomalies • B- Glaucoma associated with non- acquired systemic disease or syndrome • C- Glaucoma associated with acquired condition • D- Glaucoma following Cataract surgery 1 1/29/2018 Glaucoma associated with non- acquired ocular anomalies • Conditions with predominantly ocular anomalies present at birth which may or may not be associated with systemic signs • Axenfeld Reiger anomaly • Peters anomaly • Ectropion Uvae • Congenital iris hypolplasia • Aniridia • Oculodermal melanocytosis • Posterior polymorphous dystrophy • Microphthalmos • Microcornea • Ectopia Lentis ( et pupillae) • Persistent foetus vasculopathy Glaucoma associated with non- acquired systemic disease or syndrome predominantly associated with known syndrome, systemic anomalies present at birth which may be associated with ocular signs • Down Syndrome • Connective tissue disorder: Marfan syndrome, Weill- Marchesiani syndrome, Stickler syndrome • Metabolic disorder : Homocystenuria, lowe syndrome, Mucoploysacchroidoses • Phacomatoses: Neurofibromatoses, Sturge Weber, Klipple-Trenaunay- weber syndrome, Rubenstein Taybi • Congenital Rubella 2 1/29/2018 Glaucoma associated with acquired condition Conditions -

Expanding the Phenotypic Spectrum of PAX6 Mutations: from Congenital Cataracts to Nystagmus
G C A T T A C G G C A T genes Article Expanding the Phenotypic Spectrum of PAX6 Mutations: From Congenital Cataracts to Nystagmus Maria Nieves-Moreno 1,* , Susana Noval 1 , Jesus Peralta 1, María Palomares-Bralo 2 , Angela del Pozo 3 , Sixto Garcia-Miñaur 4, Fernando Santos-Simarro 4 and Elena Vallespin 5 1 Department of Ophthalmology, Hospital Universitario La Paz, 28046 Madrid, Spain; [email protected] (S.N.); [email protected] (J.P.) 2 Department of Molecular Developmental Disorders, Medical and Molecular Genetics Institue (INGEMM) IdiPaz, CIBERER, Hospital Universitario La Paz, 28046 Madrid, Spain; [email protected] 3 Department of Bioinformatics, Medical and Molecular Genetics Institue (INGEMM) IdiPaz, CIBERER, Hospital Universitario La Paz, 28046 Madrid, Spain; [email protected] 4 Department of Clinical Genetics, Medical and Molecular Genetics Institue (INGEMM) IdiPaz, CIBERER, Hospital Universitario La Paz, 28046 Madrid, Spain; [email protected] (S.G.-M.); [email protected] (F.S.-S.) 5 Department of Molecular Ophthalmology, Medical and Molecular Genetics Institue (INGEMM) IdiPaz, CIBERER, Hospital Universitario La Paz, 28046 Madrid, Spain; [email protected] * Correspondence: [email protected] Abstract: Background: Congenital aniridia is a complex ocular disorder, usually associated with severe visual impairment, generally caused by mutations on the PAX6 gene. The clinical phenotype of PAX6 mutations is highly variable, making the genotype–phenotype correlations difficult to establish. Methods: we describe the phenotype of eight patients from seven unrelated families Citation: Nieves-Moreno, M.; Noval, with confirmed mutations in PAX6, and very different clinical manifestations. -

WAGR Syndrome: Clinical Features and Guidelines for Management
WAGR syndrome: clinical features and guidelines for management Kelly Trout, BSN, RN International WAGR Syndrome Association 2020 Introduction WAGR syndrome is a rare multiple congenital-anomaly syndrome caused by interstitial deletion of the distal portion of chromosome 11p13. Deletion size varies between individuals from 1 million to 26.5 million base pairs, with an average of 11 million base pairs. Variability in size of the genetic deletion is thought to account for the variable phenotype. WAGR is an acronym for the most prominent features: W is for Wilms tumor, A for aniridia, G for genitourinary anomalies, and R for range of developmental delays. Wilms tumor and genital anomalies are caused by deletion of the WT1 tumor-suppressor gene, and aniridia is caused by deletion of the PAX6 ocular development gene. Developmental delays are presumed to be the result of deletion of as yet unidentified genes in the region. Most cases are identified by chromosome studies of children with isolated aniridia and are due to de novo deletions, although a few familial translocations have been reported. Individuals with WAGR syndrome have a high risk for development of Wilms tumor and late-onset renal failure, as well as a variety of additional associated conditions. Diagnosis ● Most cases of WAGR syndrome are identified in infants with isolated aniridia, 30% of whom will be positive for the characteristic deletion (11p13) ● In rare cases, aniridia may not be present. Children with Wilms tumor and genital anomalies may also warrant genetic testing ● -
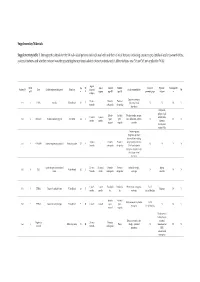
Solved/Unsolved
Supplementary Materials: Supplementary table 1. Demographic details for the 54 individual patients (solved/unsolved) and their clinical features including cataract type, details of ocular co-morbidities, systemic features and whether cataract was the presenting feature (non-isolated cataract patients only). Abbreviations: yes (Y), no (N), not applicable (N/A). Age at Famil Ag M/ Age at Cataract Cataract Cataract Systemic Consanguinit Patient ID Gene Confirmed genetic diagnosis Ethnicity diagnosi Ocular co-morbidities FH y ID e F surgery type RE type LE presenting sign features y s (days) Aniridia, nystagmus, 23 years Posterior Posterior 1-1 1 PAX6 Aniridia White British 25 F - glaucoma, foveal N N N Y 4 months subcapsular subcapsular hypoplasia Cleft palate, epilepsy, high Aphakia Aphakia Macular atrophy, myopia, 7 years 9 7 years 8 arched palate, 2-1 2 COL11A1 Stickler syndrome, type II Not Stated 34 F (post- (post- lens subluxation, vitreous N N N months months flattened surgical) surgical) anomaly maxilla, short stature (5'2ft) Anterior segment dysgenesis, pupillary abnormalities including 12 years Posterior Posterior ectopic pupils, ectropion 3-1 3 CPAMD8 Anterior segment dysgenesis 8 Other, Any other 27 F - N N Y N 5 months subcapsular subcapsular UVAE and irodensis, nystagmus, dysplastic optic discs, large corneal diameters Gyrate atrophy of choroid and 23 years 29 years 1 Posterior Posterior Retinal dystrophy, Bipolar 4-1 4 OAT White British 42 F N N N retina 7 months month subcapsular subcapsular exotropia disorder 1 year 6 1 year -

TUBB3 M323V Syndrome Presents with Infantile Nystagmus
G C A T T A C G G C A T genes Case Report TUBB3 M323V Syndrome Presents with Infantile Nystagmus Soohwa Jin 1, Sung-Eun Park 2, Dongju Won 3, Seung-Tae Lee 3, Sueng-Han Han 2 and Jinu Han 4,* 1 Department of Opthalmology, Yonsei University College of Medicine, Seoul 03722, Korea; [email protected] 2 Department of Ophthalmology, Institute of Vision Research, Severance Hospital, Yonsei University College of Medicine, Seoul 03722, Korea; [email protected] (S.-E.P.); [email protected] (S.-H.H.) 3 Department of Laboratory Medicine, Severance Hospital, Yonsei University College of Medicine, Seoul 03722, Korea; [email protected] (D.W.); [email protected] (S.-T.L.) 4 Department of Ophthalmology, Institute of Vision Research, Gangnam Severance Hospital, Yonsei University College of Medicine, Seoul 06273, Korea * Correspondence: [email protected]; Tel.: +82-2-2019-3445 Abstract: Variants in the TUBB3 gene, one of the tubulin-encoding genes, are known to cause congenital fibrosis of the extraocular muscles type 3 and/or malformations of cortical development. Herein, we report a case of a 6-month-old infant with c.967A>G:p.(M323V) variant in the TUBB3 gene, who had only infantile nystagmus without other ophthalmological abnormalities. Subsequent brain magnetic resonance imaging (MRI) revealed cortical dysplasia. Neurological examinations did not reveal gross or fine motor delay, which are inconsistent with the clinical characteristics of patients with the M323V syndrome reported so far. A protein modeling showed that the M323V mutation in the TUBB3 gene interferes with αβ heterodimer formation with the TUBA1A gene. -

MECHANISMS in ENDOCRINOLOGY: Novel Genetic Causes of Short Stature
J M Wit and others Genetics of short stature 174:4 R145–R173 Review MECHANISMS IN ENDOCRINOLOGY Novel genetic causes of short stature 1 1 2 2 Jan M Wit , Wilma Oostdijk , Monique Losekoot , Hermine A van Duyvenvoorde , Correspondence Claudia A L Ruivenkamp2 and Sarina G Kant2 should be addressed to J M Wit Departments of 1Paediatrics and 2Clinical Genetics, Leiden University Medical Center, PO Box 9600, 2300 RC Leiden, Email The Netherlands [email protected] Abstract The fast technological development, particularly single nucleotide polymorphism array, array-comparative genomic hybridization, and whole exome sequencing, has led to the discovery of many novel genetic causes of growth failure. In this review we discuss a selection of these, according to a diagnostic classification centred on the epiphyseal growth plate. We successively discuss disorders in hormone signalling, paracrine factors, matrix molecules, intracellular pathways, and fundamental cellular processes, followed by chromosomal aberrations including copy number variants (CNVs) and imprinting disorders associated with short stature. Many novel causes of GH deficiency (GHD) as part of combined pituitary hormone deficiency have been uncovered. The most frequent genetic causes of isolated GHD are GH1 and GHRHR defects, but several novel causes have recently been found, such as GHSR, RNPC3, and IFT172 mutations. Besides well-defined causes of GH insensitivity (GHR, STAT5B, IGFALS, IGF1 defects), disorders of NFkB signalling, STAT3 and IGF2 have recently been discovered. Heterozygous IGF1R defects are a relatively frequent cause of prenatal and postnatal growth retardation. TRHA mutations cause a syndromic form of short stature with elevated T3/T4 ratio. Disorders of signalling of various paracrine factors (FGFs, BMPs, WNTs, PTHrP/IHH, and CNP/NPR2) or genetic defects affecting cartilage extracellular matrix usually cause disproportionate short stature. -

Pax6 During Visual System Development
Hedgehog-dependent E3-ligase Midline1 regulates ubiquitin-mediated proteasomal degradation of Pax6 during visual system development Thorsten Pfirrmanna,1, Enrico Jandta,1, Swantje Ranfta,b, Ashwin Lokapallya, Herbert Neuhausa, Muriel Perronc, and Thomas Hollemanna,2 aInstitute for Physiological Chemistry, University of Halle-Wittenberg, 06114 Halle, Germany; bGynecological Hospital, University Medical Center Mannheim, 68167 Mannheim, Germany; and cParis-Saclay Institute of Neuroscience, CNRS, Univ Paris Sud, Université Paris-Saclay, 91405 Orsay, France Edited by Richard M. Harland, University of California, Berkeley, CA, and approved July 19, 2016 (received for review January 16, 2016) Pax6 is a key transcription factor involved in eye, brain, and pancreas remains unclear how Pax6 protein is removed from the eyestalk development. Although pax6 is expressed in the whole prospective territory on time. Some authors report the regulation of Pax6 retinal field, subsequently its expression becomes restricted to the activity by posttranslational modifications (21–23), and most optic cup by reciprocal transcriptional repression of pax6 and pax2. interestingly, Tuoc et al. showed that in cortical progenitor cells, However, it remains unclear how Pax6 protein is removed from the Pax6 protein is degraded by the proteasome mediated by Trim11 eyestalk territory on time. Here, we report that Mid1, a member of (24). However, the existence of similar mechanisms leading to the RBCC/TRIM E3 ligase family, which was first identified in patients the development of the visual system is not known. with the X-chromosome–linked Opitz BBB/G (OS) syndrome, inter- The data of our present study show that Midline1 (Mid1) acts with Pax6. We found that the forming eyestalk is a major do- serves as one of these links. -

Journal of Ophthalmology & Clinical Research
ISSN: 2573-9573 Case Report Journal of Ophthalmology & Clinical Research Bilateral Congenital Ectropion Uveae, Anterior Segment Dysgenesis and Aniridia with Microspherophakic Congenital Cataracts and RubeosisIridis Rao Muhammad Arif Khan* and Ashal Kaiser Pal *Corresponding author Rao Muhammad Arif Khan, MCPS, FCPS, FPO, FACS, Pediatric Ophthalmologist, King Edward Medical University, Al-Awali Street, Taif Road, Makkah, Saudi Arabia, Pediatric Ophthalmologist, King Edward Medical University, Tel: 00966560479694; E-mail: [email protected] Makkah, Saudi Arabia Submitted: 02 Apr 2018; Accepted: 12 Apr 2018; Published: 19 Apr 2018 Abstract In recent times, multiple eye diseases have been seen associated with an increase in the rate of Demodex infestation as a possible cause, but in the particular case of dry eye syndrome in patients treated with platelet-rich plasma, this increase in mite may be relevant to guide a more adequate treatment focusing on the elimination of the mite in conjunction with the recovery of the ocular ecology. The demodex mite is a commensal parasite that lives in hair follicles, sebaceous glands and meibomian, which in a high rate of infestation can generate alterations in the ocular area. Performing an adequate diagnosis for the detection of the mite and treatment for its eradication can be effective for the recovery of the normal physiology of the tear film that constitutes a cause of dry eye. Introduction Congenital ectropion uvea is a rare ocular manifestation of neural crest syndrome [1]. It is a non-progressive anomaly characterized by presence of iris pigment epithelium on anterior surface of iris from the pigment ruff [2]. Congenital glaucoma is its common association [3-8]. -

Lid and Lash Conditions
Perth Veterinary Ophthalmology Lid and Lash Conditions Eyelid Diseases The most common eyelid diseases are entropion, ectropion and facial droop. Entropion Entropion means a turning in of the lids. This is a common complaint in young dogs but can sometimes affect older dogs and cats as well. Most cases in young dogs affect the lower lids, but the upper lid can become affected in later life in some breeds such as Cocker Spaniels and Bloodhounds. Entropion Some breeds such as Shar Peis, Chows, Rottweillers and Mastiffs can have very complex entropion leading to defects in both upper and lower lids. A Shar Pei with severe upper and lower lid entropion Entropion is painful and can be potentially blinding. The rolling in of the lid leads to hair coming into contact with the cornea, leading to pain, ulceration and scarring (which can affect vision). In severe cases this can even lead to perforation of the eye. There are many causes of entropion. It can be primary or secondary to other problems affecting the lids (such as ectopic cilia, distichiasis etc. - see below). Some possible causes include the lid being too long, the lid being too tight, instability of the lateral canthus (outer cornea of the eyelids), misdirection of the lateral canthal tendon, brachycephalic anatomy (big eyes and short nose - e.g. Pekingese, Pugs, Shih Tsus, Persian cats etc.), diamond eye defects, loose or too much skin, facial droop etc. Often these cases are referred to a veterinary ophthalmologist for proper assessment and treatment to provide the best outcome. Entropion requires surgical correction. -

Congenital Ocular Anomalies in Newborns: a Practical Atlas
www.jpnim.com Open Access eISSN: 2281-0692 Journal of Pediatric and Neonatal Individualized Medicine 2020;9(2):e090207 doi: 10.7363/090207 Received: 2019 Jul 19; revised: 2019 Jul 23; accepted: 2019 Jul 24; published online: 2020 Sept 04 Mini Atlas Congenital ocular anomalies in newborns: a practical atlas Federico Mecarini1, Vassilios Fanos1,2, Giangiorgio Crisponi1 1Neonatal Intensive Care Unit, Azienda Ospedaliero-Universitaria Cagliari, University of Cagliari, Cagliari, Italy 2Department of Surgery, University of Cagliari, Cagliari, Italy Abstract All newborns should be examined for ocular structural abnormalities, an essential part of the newborn assessment. Early detection of congenital ocular disorders is important to begin appropriate medical or surgical therapy and to prevent visual problems and blindness, which could deeply affect a child’s life. The present review aims to describe the main congenital ocular anomalies in newborns and provide images in order to help the physician in current clinical practice. Keywords Congenital ocular anomalies, newborn, anophthalmia, microphthalmia, aniridia, iris coloboma, glaucoma, blepharoptosis, epibulbar dermoids, eyelid haemangioma, hypertelorism, hypotelorism, ankyloblepharon filiforme adnatum, dacryocystitis, dacryostenosis, blepharophimosis, chemosis, blue sclera, corneal opacity. Corresponding author Federico Mecarini, MD, Neonatal Intensive Care Unit, Azienda Ospedaliero-Universitaria Cagliari, University of Cagliari, Cagliari, Italy; tel.: (+39) 3298343193; e-mail: [email protected].