Engineering the Atomic Structure of Sequence-Defined Peptoid Polymers and Their Assemblies
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Oral Abstracts
1734-A-1902 Model systems for heterogeneous catalysts at the atomic level Hans-Joachim Freund1 1 Fritz-Haber-Institut der Max-Planck-Gesellschaft Our understanding of catalysis, and in particular heterogeneous catalysis, is to a large extent based on the investigation of model systems. Increasing the complexity of the models towards oxide supported nanoparticles, resembling a real disperse metal catalyst, allows one to catch in the model some of the important aspects that cannot be covered by metal or oxide single crystals per se. The main purpose of our studies is to provide conceptual insight into questions concerned with a variety of topics in catalysis, including support nanoparticle interaction, reactivity at the particle-support interface, strong metal support interaction, reactions in confined space and development of new instrumentation for surface science studies. The talk will address some of those issues. 1 1773-A-1902 Special metallic gasket sealing for the non-circular profile flanges Gao-Yu Hsiung1 1 NSRRC, Hsinchu, Taiwan Various types of the metallic gaskets for the non-circular profile flanges and the potential applications will be introduced. The sealing surface of flange is flat to accommodate the special metallic gasket with knife edges for the sealing. Both the flange and gasket are made of aluminum alloys and produced by the oil-free Ethanol-CNC-machining process that any non-circular profile, e. g. race-track, rectangular, key-hole, etc., flanges can be made. All the flanges and gaskets after oil-free machining can be assembled immediately without any chemical cleaning. The quality of ultrahigh vacuum at pressure < 20 nPa after vacuum baking has been approved. -

Report of Engineer's Committee on Three Mile Island
$e 0 C' 0 UNITED STATES NUCLEAR REGULATORY COMMISSION I ANNOUNCEMENT NO. 37 DATE: April 14, 1980 TO: Office Directors SUBJECT: REPORT OF ENGINEER'S COMMITTEE ON THREE MILE ISLAND The American Society of Mechanical Engineers has made available the attached report: " A REVIEW OF THE "REPORT OF THE PRESIDENT'S COMMISSION ON THE ACCIDENT AT THREE MILE ISLAND". Copies of the report are available for NRC staff from the Publications Services Unit, 016 Phillips Building. William J Besaw, Director Division of Technical Information and Document Control, ADM Attachment: As stated NRC Formq (1/76) A REVIEW OF THE "REPORT OF THE PRESIDENT'S COMMISSION ON THE ACCIDENT AT THREE MILE ISLAND" by the Engineers Committee On Three Mile Island March, 1980 Members and Alternates ENGINEERS COMMITTEE ON THREE MILE ISLAND* Peter A. Morris, Ph.D. American Nuclear Society Dwight Bellinger, P.E. - Chairman, William Ramsay, P.E. American Society for Quality Control American Society of Heating, Refrigerating and Air- S. Neal Broome, P.E. Conditioning Engineers Society of.Fire Protection Engineers John Reiter Spencer Bush, P.E., Ph.D. Society of Naval Architects American Society for Metals and Marine. Engineers Robert Cunitz, Ph.D. A.R. Robinson, P.E. System Safety Society American Society of Agricultural Engineers Brian Dunfield American Association of Coft Gavriel Salvendy, Ph.D. Engineers American Institute of Industrial Engineers Angelo Giambusso American Nuclear Society Otto A. Tennant, P.E. Vice Chairman J. Read Holland, Ph.D. National Society of Professional The Metallurgical Society of AIME Engineers Philip W. Hufnell, P.E. Harold Walton, Ph.D. -

About the Authors
1845 About the Authors Chong H. Ahn Chapter B.19 Authors University of Cincinnati Dr. Chong Ahn is a Professor of Electrical and Computer Engineering at the Department of Electrical University of Cincinnati. He obtained his Ph.D. degree in Electrical Engineering from and Computer Engineering the Georgia Institute of Technology in 1993 and then worked as a postdoctoral fellow and Computer Science at IBM T.J. Watson Research Center. His research interests include all aspects of Cincinnati, OH, USA [email protected] design, fabrication, and characterization of magnetic MEMS devices, microfluidic devices, protein chips, lab-on-a-chips, nano biosensors, point-of-care testing and BioMEMS systems. He is an associate editor of the IEEE Sensors Journal. Boris Anczykowski Chapter C.27 nanoAnalytics GmbH Dr. Boris Anczykowski is a physicist with an extensive research background in the Münster, Germany field of dynamic Scanning Force Microscopy. He co-invented the Q-Control technique [email protected] and received the Innovation Award Münsterland for Science and Economy in 2001 for this achievement. He is a managing director and co-founder of nanoAnalytics GmbH, a company specialized in the characterization of surfaces and interfaces on the micro- and nanometer scale. Massood Z. Atashbar Chapter A.7 Western Michigan University Professor Massood Z. Atashbar received the B.Sc. degree in electrical Department of Electrical engineering from the Isfahan University of Technology, Tehran, Iran, and Computer Engineering the M.Sc. degree in electrical engineering from the Sharif University of Kalamazoo, MI, USA Technology, Tehran, and the Ph.D. degree from the Department of [email protected] Communication and Electronic Engineering, RMIT University, Melbourne, Australia, in 1998. -

International Conference on Nuclear Knowledge Management: Strategies, Information Management and Human Resource Development
Flyer.qxd 2004-03-24 09:00 Page 1 International Conference on Steering Committee Objectives Contact Information Nuclear Knowledge Management: International Atomic H.S. Cherif The objective of this conference is to reach a clear and com- International Atomic Energy Agency Energy Agency P.J. Gowin mon understanding of issues related to nuclear knowledge Conference IAEA - CN-123 Strategies, Information Y. Ya n ev management for sustaining knowledge and expertise in nuclear Vienna International Centre France, CEA B. Gillet science and technology. Wagramer Strasse 5 CEA/INSTN D. Gentile Management and Human P.O.Box 100 CEA/INSTN C. Feltin The conference will provide a forum for professionals and deci- A-1400 Vienna, Austria EUSIDIC J. van Halm Resource Development European Commission G. van Goethem sion makers in the nuclear sector, comprising industry, govern- Tel.: +43 1 26000 FORATOM P. Haug ments and academia as well as professionals in the knowledge Fax: +43 1 26007 Japan Atomic Industrial Forum N. Ishizuka management and information technology sectors E-mail: [email protected] OECD/Nuclear Energy Agency T. Haapalehto World Nuclear University J. Ritch, World Nuclear Association ∑ to exchange information and share experience on nuclear WONUC A. Maïsseu knowledge management, comprising strategies, informa- Scientific Secretariat (IAEA) Argentina, Atomic R. Cirimello tion management and human resource development; H.S. Cherif Department of Management Energy Commission P.J. Gowin Department of Nuclear Energy Belgium, Nuclear Research F. Moons ∑ to identify lessons learned and to embark on the develop- Y.L. Yanev Department of Nuclear Energy Centre (SCK•CEN) ment of new initiatives and concepts for nuclear knowl- France, CEA C. -

RESEARCH STATEMENT: My Current Research Interests Lie in the Intersection of Novel III-V Materials, Hetero-Structures, and Photonic and Electronic Quantum Devices
Department of Electrical and Computer Engineering, Prof. Can Bayram University of Illinois at Urbana-Champaign, IL, USA PROFESSIONAL POSITIONS: 2020 – present Assoc. Prof. (tenured), Electrical and Computer Engineering Dept., Resident Faculty, Nick Holonyak, Jr. Micro and Nanotechnology Laboratory, Affiliate Faculty, Carle Illinois College of Medicine, Engineering Partner, Carle Illinois College of Medicine, University of Illinois at Urbana-Champaign, IL, USA 2014 – 2020 Asst. Prof. (tenure-track), Electrical and Computer Engineering Dept., Resident Faculty, Nick Holonyak, Jr. Micro and Nanotechnology Laboratory, University of Illinois at Urbana-Champaign, IL, USA 2011 – 2014 Postdoctoral Research Scientist, IBM Research, Thomas J. Watson Research Center, Yorktown Heights, NY, USA EDUCATION: 2005 – 2011 Ph.D. (Electrical Engineering) Northwestern University, Illinois, USA Ph.D. focus on Solid State Devices and Photonics 2001 – 2005 B.S. (Electrical Engineering) Bilkent University, Ankara, TURKEY B.S. focus on Physical Electronics RESEARCH STATEMENT: My current research interests lie in the intersection of novel III-V materials, hetero-structures, and photonic and electronic quantum devices. Particularly, my research group explores III-V materials and novel devices, hetero-integration of III-Vs on unconventional platforms such as graphene and silicon, heat transport across/through semiconductors, efficiency droop mechanisms and remedies in AlInGaN emitters, and ultra- fast THz photonics/electronics. My work is interdisciplinary. My team -

Infrared and Ultra-Violet Regions
CQD Atomic Engineering of III-V Semiconductors for optoelectronic Quantum Devices from; Deep UV(200nm) to THZ( 300 microns) Manijeh Razeghi Center for Quantum Devices Dept. of Electrical Engineering and Computer Science Northwestern University, Evanston, IL 60208 email: [email protected], Ph: (847) 491-7251, Fax: (847) 467-1817 Boston Chapter of IEEE Photonics Society: Emerging Optical Materials Workshop Wednesday, April 12, 2017 1 Major Achievements of 20th Century CQD Physical Science Q-mechanics Atomic Structure •Semiconductors •Superconductors •Simulation •Polymers •Interconnection Natural Information Electronics Science Science Computer& Internet •Artificial Neural •Simulation Networks •Interconnection •Fuzzy Logic Life Science Genetics Gene & Cell Structure 2 Quantum Optoelectronic Devices and ResearchCQD optoelectronic devices will be involved in all areas of a human’s life, trying to improve the functionality of its body and mind. Examples of Quantum device applications are given below. EXPLORATION (Space & Underwater) needs reliable electronics in all these domains • Pollution sensors • Food safety • Solar Cells • ... • Energy efficient • Virtual reality • Bullet medicine devices (LEDs...) Food • ... • Sensors • Biomimetrics Energy Health • ... Medicine Needs of each individual in Society Entertainment Transportation • Virtual reality Communication • Computers • Electrical vehicles • Optical fiber (WDM, DWDM) • Video games • Computers • Wireless • Interactive media • Sensors • Personal Digital Assistant • Internet -

Challenges for Decommissioning Policies
IAEA SAFETY RELATED PUBLICATIONS IAEA SAFETY STANDARDS Under the terms of Article III of its Statute, the IAEA is authorized to establish or adopt standards of safety for protection of health and minimization of danger to life and property, and to provide for the application of these standards. The publications by means of which the IAEA establishes standards are issued in the IAEA Safety Standards Series. This series covers nuclear safety, radiation safety, transport safety and waste safety, and also general safety (i.e. all these areas of safety). The publication categories in the series are Safety Fundamentals, Safety Requirements and Safety Guides. Safety standards are coded according to their coverage: nuclear safety (NS), radiation safety (RS), transport safety (TS), waste safety (WS) and general safety (GS). Information on the IAEA’s safety standards programme is available at the IAEA Internet site http://www-ns.iaea.org/standards/ The site provides the texts in English of published and draft safety standards. The texts of safety standards issued in Arabic, Chinese, French, Russian and Spanish, the IAEA Safety Glossary and a status report for safety standards under development are also available. For further information, please contact the IAEA at P.O. Box 100, 1400 Vienna, Austria. All users of IAEA safety standards are invited to inform the IAEA of experience in their use (e.g. as a basis for national regulations, for safety reviews and for training courses) for the purpose of ensuring that they continue to meet users’ needs. Information may be provided via the IAEA Internet site or by post, as above, or by email to [email protected]. -

Advanced Components for Electric and Hybrid Electric Vehicles
M m III Hi 1 MIST ^^^^^^1 jljlll 1 iV PUBLICATIONS A11104 EfifilfiT United States Department of Commerce Technology Administration National Institute of Standards and Technology NIST Special Publication 860 Advanced Components for Electric and Hybrid Electric Vehicles Workshop Proceedings October 27-28, 1993 Gaithersburg, Maryland K. L. Stricklett, Editor 7he National Institute of Standards and Technology was established in 1988 by Congress to "assist industry in the development of technology . needed to improve product quality, to modernize manufacturing processes, to ensure product reliability . and to facilitate rapid commercialization ... of products based on new scientific discoveries." NIST, originally founded as the National Bureau of Standards in 1901, works to strengthen U.S. industry's competitiveness; advance science and engineering; and improve public health, safety, and the environment. One of the agency's basic functions is to develop, maintain, and retain custody of the national standards of measurement, and provide the means and methods for comparing standards used in science, engineering, manufacturing, commerce, industry, and education with the standards adopted or recognized by the Federal Government. As an agency of the U.S. Commerce Department's Technology Administration, NIST conducts basic and applied research in the physical sciences and engineering and performs related services. The Institute does generic and precompetitive work on new and advanced technologies. NIST's research facilities are located at Gaithersburg, -

Selective Guide to Literature on Nuclear Engineering
Engineering Literature Guides, Number 27 Selective Guide to Literature on Nuclear Engineering Compiled by Theresa S. Lee Media Union Library University of Michigan Ann Arbor, Michigan February 2000 American Society for Engineering Education Engineering Libraries Division 1818 N Street, NW, Suite 600 Washington, D.C. 20036 Copyright © 2000 by American Society for Engineering Education 1818 N.W., Suite 600 Washington, DC 20036-2479 International Standard Book Number: 0-87823-183-8 Manufactured in the United States of America Godlind Johnson ELD Publications Committee Chair Engineering Library SUNY-Stony Brook Stony Brook, NY 11794 Table of Contents Introduction ...................................................................................................................1 Guides to the Literature...................................................................................................2 Printed and Electronic Indexes and Abstracts ...............................................................3 Dictionaries.................................................................................................................... 6 Encyclopedias.................................................................................................................7 Handbooks and Tables ..................................................................................................8 Directories Nuclear Industries and Organizations ........................................................12 Biographical ...............................................................................................14 -
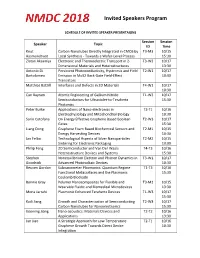
Invited Speakers Program
Invited Speakers Program SCHEDULE OF INVITED SPEAKER PRESENTAIONS Session Session Speaker Topic ID Time Knut Carbon Nanotubes Directly Integrated in CMOS by T3-M3 10/15 Aasmundtveit Local Synthesis - Towards a Wafer-Level Process 15:30 Zlatan Aksamija Electronic and Thermoelectric Transport in 2- T3-W2 10/17 Dimensional Materials and Heterostructures 13:30 Antonio Di Persistent Photoconductivity, Hysteresis and Field T2-W1 10/17 Bartolomeo Emission in MoS2 Back-Gate Field-Effect 10:30 Transistors Matthias Batzill Interfaces and Defects in 2D Materials T4-W1 10/17 10:30 Can Bayram Atomic Engineering of Gallium Nitride T1-W3 10/17 Semiconductors for Ultraviolet-to-Terahertz 15:30 Photonics Peter Burke Applications of Nano-electronics in T3-T1 10/16 Electrophysiology and Mitochondrial Biology 10:30 Sorin Cotofana On Energy Effective Graphene Based Boolean T2-W3 10/17 Gates 15:30 Liang Dong Graphene Foam Based Biochemical Sensors and T2-M1 10/15 Energy Harvesting Devices 10:30 Jan Felba Technological Aspects of Silver Nanoparticles T2-M2 10/15 Sintering for Electronic Packaging 13:30 Philip Feng 2D Semiconductor and Van Der Waals T4-T3 10/16 Heterostructure Devices and Systems 15:30 Stephen Nonequilibrium Electron and Phonon Dynamics in T3-W1 10/17 Goodnick Advanced Photovoltaic Devices 10:30 Reuven Gordon Subnanometer Plasmonics: Quantum Regime T1-T3 10/16 Functional Metasurfaces and the Plasmonic 15:30 Coulomb Blockade Bonnie Gray Polymer Nanocomposites for Flexible and T3-M2 10/15 Wearable Fluidic and Biomedical Microdevices 13:30 Mona -

Conference Book
GSELOP2021 Global Summit and Expo on Laser, Optics and Photonics August 23-25, 2021 Paris, France The Scientistt Bangalore, Karnataka, India Contact: +91 77 99 83 5553 Email: [email protected] GSELOP2021 Global Summit and Expo on Laser, Optics and Photonics August 23-25, 2021 | Paris, France FOREWORD Dear Colleagues, We are pleased to announce that the Global Summit and Expo on Laser, Optics and Photonics(GSELOP2021) will be held during August 23-25, 2021 in Paris, France is a premiere and one of the highest level international academic conferences in the field of laser, optics and photonics. The GSELOP2021 will present the most recent advances in technology developments and business opportunities in laser, optics and photonics commercialization. Highly cited researchers from renowned universities across the globe and industry leaders will share their research and vision, while selected talks from industrial exhibitors will present commercial showcases in all current market fields of optics and photonics. This conference offers an excellent forum for the state of art presentations by invited speakers, leading specialists in the field of laser, optics and photonics. Most recent developments, progress and achievements realized in the fields covered by the conference will be presented in plenary, keynote presentations and short oral contributions as well as in poster sessions. As you enjoy the intellectual interaction with peers and leaders in the field, we encourage you to immerse yourselves in the Paris experience with her rich cultural diversity. We are confident that your stay with us will be enriching and fascinating. Welcome to Paris and we wish you a pleasant, fruitful and unforgettable experience! Sincerely, Prof. -

October 29-November 1, 2006
Surface and Interface Science at the Atomic Scale 2006 Program Meeting Airlie Conference Center Warrenton, Virginia October 29 - November 1, 2006 Division of Materials Sciences and Engineering Cover Top left: A SrTiO3 bicrystal grain boundary intersecting the (100) surface which is in a R5xR5 reconstruction (see enlargement). The surface potential at the boundary is imaged directly with scanning surface potential microscopy. [courtesy Dawn Bonnell, U. Penn.] Top right: Atomic resolution Z-contrast image of a Si3N4 grain, oriented along the [0001] zone axis, with a hexagonal edge showing the interface with Lu2O3. Si, N and Lu atoms are shown schematically in white, gray and blue circles, respectively. [courtesy N. D. Browning, UC Davis and LLNL, J.-C. Idrobo, ORNL and Vanderbilt, and A. Ziegler, Max-Planck Institute for Biochemistry] Center left: The circular quantum corral and the linear electron resonator are built using individual silver atoms extracted locally with a scanning tunneling microscope tip manipulation on a Ag(111) surface at liquid helium temperatures. The circular corral is used for the investigations of detailed atom movement mechanisms during single atom manipulation. The linear electron resonator is used for the transport of sexiphenyl molecules. Here, the two sexiphenyl molecules are positioned at the left end of the resonator to shoot them toward two target atoms located at the right end using scanning tunneling microscope tip. [Courtesy: Saw-Wai Hla, Ohio University] Center right: Structure of a catalytic Ti-Al surface complex predicted by first-principles theory to be highly active in the dissociative chemisorption of molecular hydrogen (H2), a key step in hydrogen storage reactions.