Cloning, Sequence Analysis, and Characterization of the Lysyl Oxidase
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Page Numbers in Bold Indicate Main Discus- Sion of Topic. Page Numbers
168397_P489-520.qxd7.0:34 Index 6-2-04 26p 2010.4.5 10:03 AM Page 489 source of, 109, 109f pairing with thymine, 396f, 397, 398f in tricarboxylic acid cycle, 109–111, 109f Adenine arabinoside (vidarabine, araA), 409 Acetyl CoA-ACP acetyltransferase, 184 Adenine phosphoribosyltransferase (APRT), Index Acetyl CoA carboxylase, 183, 185f, 190 296, 296f in absorptive/fed state, 324 Adenosine deaminase (ADA), 299 allosteric activation of, 183–184, 184f deficiency of, 298, 300f, 301–302 allosteric inactivation of, 183, 184f gene therapy for, 485, 486f dephosphorylation of, 184 Adenosine diphosphate (ADP) in fasting, 330 in ATP synthesis, 73, 77–78, 78f Page numbers in bold indicate main discus- hormonal regulation of, 184, 184f isocitrate dehydrogenase activation by, sion of topic. Page numbers followed by f long-term regulation of, 184 112 denote figures. “See” cross-references direct phosphorylation of, 183–184 transport of, to inner mitochondrial short-term regulation of, 183–184, 184f membrane, 79 the reader to the synonymous term. “See Acetyl CoA carboxylase-2 (ACC2), 191 in tricarboxylic acid cycle regulation, 114, also” cross-references direct the reader to N4-Acetylcytosine, 292f 114f related topics. [Note: Positional and configura- N-Acetyl-D-glucosamine, 142 in urea cycle, 255–256 N-Acetylgalactosamine (GalNAc), 160, 168 ribosylation, 95 tional designations in chemical names (for N-Acetylglucosamindase deficiency, 164f Adenosine monophosphate (AMP; also called example, “3-“, “α”, “N-“, “D-“) are ignored in N-Acetylglucosamine (GlcNAc), -

Review Article
REVIEW ARTICLE COLLAGEN METABOLISM COLLAGEN METABOLISM Types of Collagen 228 Structure of Collagen Molecules 230 Synthesis and Processing of Procollagen Polypeptides 232 Transcription and Translation 233 Posttranslational Modifications 233 Extracellular Processing of Procollagen and Collagen Fibrillogenesis 240 Functions of Collagen in Connective rissue 243 Collagen Degradation 245 Regulation of the Metabolism of Collagen 246 Heritable Diseases of Collagen 247 Recessive Dermatosparaxis 248 Recessive Forms of EDS 251 EDS VI 251 EDS VII 252 EDS V 252 Lysyl Oxidase Deficiency in the Mouse 253 X-Linked Cutis Laxa 253 Menke's Kinky Hair Syndrome 253 Homocystinuria 254 EDS IV 254 Dominant Forms of EDS 254 Dominant Collagen Packing Defect I 255 Dominant and Recessive Forms of Osteogenesis Imperfecta 258 Dominant and Recessive Forms of Cutis Laxa 258 The Marfan Syndrome 259 Acquired Diseases and Repair Processes Affecting Collagen 259 Acquired Changes in the Types of Collagen Synthesis 260 Acquired Changes in Amounts of Collagen Synthesized 263 Acquired Changes in Hydroxylation of Proline and Lysine 264 Acquired Changes in Collagen Cross-Links 265 Acquired Defects in Collagen Degradation 267 Conclusion 267 Bibliography 267 Collagen Metabolism A Comparison of Diseases of Collagen and Diseases Affecting Collagen Ronald R. Minor, VMD, PhD COLLAGEN CONSTITUTES approximately one third of the body's total protein, and changes in synthesis and/or degradation of colla- gen occur in nearly every disease process. There are also a number of newly described specific diseases of collagen in both man and domestic animals. Thus, an understanding of the synthesis, deposition, and turnover of collagen is important for the pathologist, the clinician, and the basic scientist alike. -

Golden Yearbook
Golden Yearbook Golden Yearbook Stories from graduates of the 1930s to the 1960s Foreword from the Vice-Chancellor and Principal ���������������������������������������������������������5 Message from the Chancellor ��������������������������������7 — Timeline of significant events at the University of Sydney �������������������������������������8 — The 1930s The Great Depression ������������������������������������������ 13 Graduates of the 1930s ���������������������������������������� 14 — The 1940s Australia at war ��������������������������������������������������� 21 Graduates of the 1940s ����������������������������������������22 — The 1950s Populate or perish ���������������������������������������������� 47 Graduates of the 1950s ����������������������������������������48 — The 1960s Activism and protest ������������������������������������������155 Graduates of the 1960s ���������������������������������������156 — What will tomorrow bring? ��������������������������������� 247 The University of Sydney today ���������������������������248 — Index ����������������������������������������������������������������250 Glossary ����������������������������������������������������������� 252 Produced by Marketing and Communications, the University of Sydney, December 2016. Disclaimer: The content of this publication includes edited versions of original contributions by University of Sydney alumni and relevant associated content produced by the University. The views and opinions expressed are those of the alumni contributors and do -

FROM the DIRECTOR: OUR NUMBERS ARE up in This Issue: Scientists Are Accustomed to Dealing with Some Pretty Big Numbers – and Some Very Small Ones Too
Australian Synchrotron Lightspeed: November 2008 FROM THE DIRECTOR: OUR NUMBERS ARE UP In this issue: Scientists are accustomed to dealing with some pretty big numbers – and some very small ones too. • From the Director • A Tribute to Hans Freeman Take an electron, for example. It weighs around • Up to Speed • Beamtime Applications 9.10938188 x 10‐31 kilograms when it’s just sitting • Synchrotron opens up around. Now take a few more, turn them into a • In science, a lot can happen in one night beam 0.06 millimetres wide, and accelerate the • Experimenting with SAXS beam to say 99.9987% of the speed of light, which • One in a thousand is 299,792,458 metres per second. Then persuade • The next generation of synchrotron PhDs your electron beam to turn enough corners to keep • Another high-performance it circulating in a synchrotron storage ring 24 hours machine • Beamline Focus a day. • Rapid Access on Trial • New Survey to Boost User That’s the amazing feat regularly performed by the Satisfaction Australia Synchrotron’s team of engineers, • A traveller’s tale accelerator physicists, operators, technicians and • Events Diary • Careers at The Australian Prof. Robert Lamb other support staff. Impressed? I certainly am. Especially when that’s combined with a beam availability of 98.3 per cent that puts us in the world’s top five synchrotrons in terms of performance. And it seems our users are impressed too. Since we opened in July 2007, over 500 users have collectively visited us more than 1000 times, and we were only running at 30 per cent capacity. -
Copyright and Use of This Thesis This Thesis Must Be Used in Accordance with the Provisions of the Copyright Act 1968
COPYRIGHT AND USE OF THIS THESIS This thesis must be used in accordance with the provisions of the Copyright Act 1968. Reproduction of material protected by copyright may be an infringement of copyright and copyright owners may be entitled to take legal action against persons who infringe their copyright. Section 51 (2) of the Copyright Act permits an authorized officer of a university library or archives to provide a copy (by communication or otherwise) of an unpublished thesis kept in the library or archives, to a person who satisfies the authorized officer that he or she requires the reproduction for the purposes of research or study. The Copyright Act grants the creator of a work a number of moral rights, specifically the right of attribution, the right against false attribution and the right of integrity. You may infringe the author’s moral rights if you: - fail to acknowledge the author of this thesis if you quote sections from the work - attribute this thesis to another author - subject this thesis to derogatory treatment which may prejudice the author’s reputation For further information contact the University’s Director of Copyright Services sydney.edu.au/copyright Advanced NMR Spectral Characteristics Used in Biochemical Studies of Anisotropic Media and Cells New Ways of Analysing Quadrupolar-Split Resonances, Dynamically Hyperpolarised Spectra, and Those Obtained With Shift Reagents Max Puckeridge, BSc(Hons) A thesis submitted for the degree of Doctor of Philosophy January 2014 School of Molecular Bioscience University of Sydney NSW 2006 Australia i Acknowledgements I wish to thank my supervisor, Professor Philip Kuchel, for the large role he played in my development as a scientist and a person. -

Organic Chemistry to Biochemistry
page 1 Metabolisn1 Review: Step by Step from Organic Chemistry to Biochemistry Overview: This handout contains a review of the fundamental parts of organic chemistry needed for metabolism. Dr. Richard Feinman Department of Biochemistry Room 7-20, BSB (718) 270-2252 [email protected] STEP-BY-STEP: ORGANIC CHEMISTRY TO NUTRITION AND METABOLISM page 2 CHAPTER O. INTRODUCTION Where we're going. The big picture in nutrition and metabolism is shown in a block diagram or "black box" diagram. A black box approach shows inputs and outputs to a process that may not be understood. It is favored by engineers who are the group that are most uncomfortable with the idea that they don't know anything at all. The black box approach can frequently give ----c02 you some insight because it organizes whatever you do know. For example: ENERGY CELL MA TERIA L 1. Even though the block diagram is very simple, just looking at the inputs and outputs gives us some useful information. il The diagram says animals obtain energy by the oxidation of food to CO2 and water. Although you knew this before, the diagram highlights the fact that understanding biochemistry probably involves understanding oxidation-reduction reactions. 2. The diagram also indicates what might not be obvious: a major part of the energy obtained from oxidation of food is used to make new cell material. Although we think of organisms using energy for locomotion or to do physical work, in fact, most of the energy used is chemical energy. 3. Inside the black boxes in the diagram contain are the (organic) chemical reactions that convert food into energy and cell material. -
9781920898359 Appendices R
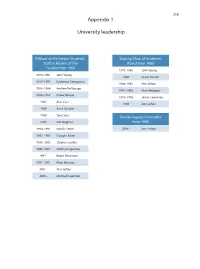
319 Appendix 1 University leadership Fellows of the Senate Students, Deputy Chair of Academic Staff or Alumni of the Board from 1980 Faculty from 1980 1978–1980 John Young 1978–1981 John Young 1986 Susan Dorsch 1979–1993 Katherine Georgouras 1986–1987 Ann Sefton 1983–1986 Andrew Refshauge 1991–1993 Alan Pettigrew 1986–1987 Diana Temple 1992–1996 James Lawrence 1987 Alan Cass 1996 Ann Sefton 1989 Anna Donald 1989 Tony Sara Senate Deputy Chancellor 1989 Eric Wegman from 1980 1990–1991 Natalie Smith 2004– Ann Sefton 1993–1995 Douglas Baird 1995–2002 Stephen Leeder 1996–2001 Michael Copeman 1997– Robin Fitzsimons 1997–2001 Peter Burrows 2001– Ann Sefton 2005– Michael Copeman 320 Appendix 2 Deans of the Faculty of Medicine from 1980 1974 –1989 Richard S Gye 1989 –1996 John Atherton Young 1997–2002 Stephen Leeder 2003 – Andrew Coats Appendix 3 Current Emeritus Professors Barry Baker John Little Tony Basten William McCarthy Geoffrey Berry James McLeod Charles Blackburn Russell Meares Charles Bridges-Webb Gerald Milton Neil Buchanan Kim Oates Peter Castaldi Murray Pheils John Chalmers Wai-On Phoon Patrick De Burgh Thomas Reeve Susan Dorsch Douglas Saunders Hans Freeman Ann Sefton Kerry Goulston Ainslie Sheil Richard Gye Frederick Stephens Akos Gyory Michael Taylor Noel Hush Thomas Taylor Gordon Johnson Stewart Truswell Charles Kerr John Turtle James Lawrence Robert Wake 321 Appendix 4 Professors appointed or promoted 1980–2005 1980 W Cramond 1989 Nicholas Hunt 1994 Leigh Delbridge 1980 Graeme Johnston 1989 John Sutton 1994 Ian Frazer 1980 Philip -

Elastin and the Lung
Thorax: first published as 10.1136/thx.41.8.577 on 1 August 1986. Downloaded from Thorax 1986;41:577-585 Review article Elastin and the lung It is essential that the framework of all multicellular most prominently displayed in newly synthesised or organisms should include some materials with high juvenile elastin, is a glycoprotein that stains with ura- tensile strength and rigidity, such as bone and col- nyl acetate and leads, which appears as small fibrils lagen, to maintain shape and mechanical rigidity. In 10-12 nm in diameter concentrated around the addition, there is a requirement for a component with periphery of the amorphous elastin. The microfibrils intrinsic elasticity that can stretch and undergo elastic are chemically and morphologically quite distinct recoil when required. This property is supplied by an from the amorphous elastin. There is some evidence unusual fibrous protein, which over 150 years ago was to suggest that the microfibrils are secreted into the given the name elastin. extracellular matrix before elastin synthesis and func- Elastin fibres are present in virtually all vertebrate tion as a nucleation site for future elastin deposition. tissues, although it is only within a few, such as ar- For more details on every aspect of the microfibrils, teries, some ligaments, and the lung, that elastin com- readers are referred to other articles.24 prises an appreciable percentage of the total protein. The purification of elastin depends predominantly The ligamentum nuchae of grazing animals and the on its remarkable insolubility, even under harsh, aorta of most vertebrates contain over 50% elastin on denaturing conditions. -

Pharmaceutical Preparation for Preventing and Treating Progressive Myopia
(19) TZZ¥_Z¥ T (11) EP 3 103 446 A2 (12) EUROPEAN PATENT APPLICATION published in accordance with Art. 153(4) EPC (43) Date of publication: (51) Int Cl.: 14.12.2016 Bulletin 2016/50 A61K 31/13 (2006.01) A61K 9/08 (2006.01) A61P 27/10 (2006.01) (21) Application number: 14872234.1 (86) International application number: (22) Date of filing: 16.12.2014 PCT/RU2014/000949 (87) International publication number: WO 2015/094019 (25.06.2015 Gazette 2015/25) (84) Designated Contracting States: • Korigodskiy, Aleksandr Robertovich AL AT BE BG CH CY CZ DE DK EE ES FI FR GB Moscow 107113 (RU) GR HR HU IE IS IT LI LT LU LV MC MK MT NL NO PL PT RO RS SE SI SK SM TR (72) Inventors: Designated Extension States: • Zakharov, Ivan Dmitrievich BA ME Moscow 109125 (RU) • Korigodskiy, Aleksandr Robertovich (30) Priority: 17.12.2013 RU 2013155972 Moscow 107113 (RU) (71) Applicants: (74) Representative: Chas. Hude A/S • Zakharov, Ivan Dmitrievich H.C. Andersens Boulevard 33 Moscow 109125 (RU) 1780 Copenhagen V (DK) (54) PHARMACEUTICAL PREPARATION FOR PREVENTING AND TREATING PROGRESSIVE MYOPIA (57) The invention relates to medicine, and more par- during the treatment of progressive myopia. These aims ticularly to opthalmology, and is intended for strengthen- are achieved through the creation and use of a pharma- ing the sclera for the purposes of preventing and treating ceutical preparation in the form of an instillable aqueous progressive myopia. The invention is directed toward solution or an eye gel or a medicated eye film containing strengthening the sclera in the case of progressive my- an effective amount of an amine with functional groups opia, reducing the time taken to strengthen the sclera, in the form of a salt or a part of a transition metal complex and preventing damaging, toxic and inflammatory effects compound or a combination thereof. -

Print This Article
Liversidge Research Lecture No. 21 1978 ELEGANCE IN MOLECULAR DESIGN: THE COPPER SITE OF PHOTOSYNTHETIC ELECTRON-TRANSFER PROTEIN HANS C. FREEMAN The Royal Society of New South Wales Liversidge Research Lecture No. 21, 1978 Royal Society of NSW 1 Hans Charles Freeman This photograph is reproduced by permission of the Australian Journal of Chemistry and the RACI 472 2 Royal Society of NSW Liversidge Research Lecture No. 21, 1978 Liversidge Research Lecturer No. 21, 1978. HANS CHARLES FREEMAN 1929-2008 Hans Charles Freeman was born on 26 May 1929 in Breslau, Germany. His family arrived in Sydney as refugees in 1938. After attending Double Bay Public School, he received his secondary education at Sydney Boys' High School where he was taught science by the ‘now-legendary’ teacher Leonard A. Basser. He proceeded to the University of Sydney, graduating B.Sc.(1st. Class Hons.), with the University Medal in 1950. While holding appointments as Teaching Fellow and Temporary Lecturer, he carried out research on the dipole moments of organic molecules in the gas state under the supervision of Professor R.J.W. Le Fèvre, and graduated M.Sc. in 1952. In 1952-3 he was a Rotary Foundation Fellow at the California Institute of Technology. There, at the suggestion of Linus Pauling, he began research in X-ray crystal structure analysis with one of the leading thinkers and strategists of the subject, Edward W. Hughes. In 1954 he returned to the University of Sydney as a Lecturer in Chemistry, and completion of the research begun in California led to the award of a Sydney Ph.D. -

United States Patent 19 11 Patent Number: 5,652,112 Eyre 45 Date of Patent: Jul
US005652112A United States Patent 19 11 Patent Number: 5,652,112 Eyre 45 Date of Patent: Jul. 29, 1997 54 ASSAY FORTYPE I COLLAGEN CARBOXY. Baldwin, et al., "Structure of cDNA clones coding for TERMINAL TELOPEPTDEANALYTES human type II procollagen; the a1(II) chain is more similar to the a1 (I) chain than two other a chains of fibrillar (75) Inventor: David R. Eyre, Mercer Island, Wash. collagens.” Biochemistry Journal, 262:521-528 (1989). Ala-Kokko, et al., "Structure of cDNA clones coding for the 73) Assignee: Washington Research Foundation, entire preproal(II) chain of human type III procollagen: Seattle, Wash. Differences in protein structure from type I procollagen and conservation of codon preferences,” Biochemistry Journal, 21 Appl. No.: 771,219 260:509-516 (1989). Seibel, “Komponentender extrazllularen Gewebematrix als 22 Filed: Dec. 20, 1996 potentielle Marker des Bindegewebs-, Knorbel-und Knochenmetabolismus bei Erkrankungen des Bewegung Related U.S. Application Data sapparates.” Zeitschrift fur Rheumatologie, 48:6-18 (1989). 63 Continuation of Ser. No. 537,502, Oct. 2, 1995, which is a Dodge and Poole, "Immunlohistochemical Detection and continuation of Ser. No. 226,070, Apr. 11, 1994, Pat. No. Immunochemical Analysis of Type II Collagen Degradation 5,455,179, which is a continuation of Ser. No. 823,270, Jan. in Human Normal, Rheumatoid, and Osteoarthritic Articular 16, 1992, Pat No. 5,532,169, which is a division of Ser. No. Cartilages and in Explants of Bovine Articular Cartilage 444,881, Dec. 1, 1989, Pat No. 5,140,103, which is a continuation-in-part of Ser. No. 118,234, Nov. -

THE AMINO ACID SEQUENCE of the CARBOXYTERMINAL NONHELICAL CROSS LINK REGION of the Al CHAIN of CALF SKIN COLLAGEN
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Volume 21, number 1 FEBS LETTERS March 1972 THE AMINO ACID SEQUENCE OF THE CARBOXYTERMINAL NONHELICAL CROSS LINK REGION OF THE al CHAIN OF CALF SKIN COLLAGEN J. RAUTERBERG, P. FIETZEK, F. REXRODT, U. BECKER, M. STARK and K. KUHN Max-Planck-lnstitut fiir Eiweiss- und Lederforschung. Abteilung Kiihn, D-8 Miinchen 2, Schillerstrasse 46, W. Germany Received 6 December 197 1 1. Introduction by centrifugation for 30 min at 15,000 g (rotor GSA) in an RC-2 Sorvall centrifuge. The supernatant was The collagen molecule terminates at both ends in dialysed against 0.06 M sodium acetate buffer pH 4.8. regions endowed with particular properties. These The arl chains were isolated on carboxy methyl regions cannot participate in the triple helical con- cellulose as described earlier [ Ill. formation since glycine does not occupy every third position as in the central areas of the molecule. The 2.2. Preparation of al-CB6 terminal regions contain lysine residues usually con- The al-chains were cleaved with CNBr and then verted into cw-amino-adipic-semialdehyde by lysine crl-CB6 was isolated using the procedure described oxidase [ 11 . Allysine is the essential participant in earlier [5] with the following modification. After the crosslinking reaction [2,3]. Structure and func- digestion, CNBr was removed by desalting on Bio- tion of the N-terminal region have been determined Gel P-2 (100-200 mesh) in acetic acid. The peptide some time ago [4-91.