View and Meta-Analysis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Neural Mechanisms of Oxytocin and Serotonin Interaction in Non-Human Primates and Patients with Autism Arthur Lefevre
Neural mechanisms of oxytocin and serotonin interaction in non-human primates and patients with autism Arthur Lefevre To cite this version: Arthur Lefevre. Neural mechanisms of oxytocin and serotonin interaction in non-human primates and patients with autism. Neuroscience. Université de Lyon, 2016. English. NNT : 2016LYSE1323. tel-01508642 HAL Id: tel-01508642 https://tel.archives-ouvertes.fr/tel-01508642 Submitted on 14 Apr 2017 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. N°d’ordre NNT : xxx THESE de DOCTORAT DE L’UNIVERSITE DE LYON opérée au sein de l’Université Claude Bernard Lyon 1 Ecole Doctorale 476 (Neuroscience et Cognition) Spécialité de doctorat : Discipline : Neurosciences Soutenue publiquement le 13 Décembre 2016, par : Arthur LEFEVRE __________________________________________ Neural mechanisms of oxytocin and serotonin interaction in non- human primates and patients with autism __________________________________________ Thèse dirigée par Angela Sirigu (DRCE, CNRS) Devant le jury composé de : GERVAIS Rémi, Président, (Professeur à l’Université Lyon 1 Claude Bernard) CHINI BICE, Rapporteur, (Senior researcher at the Institute of Neuroscience, Milan) CHAKRABARTI Bhismadev, Rapporteur, (Associate Professor at university of Reading and Senior researcher at Cambridge university) KRAUS Christoph, Examinateur, (Senior researcher at the Medical university of Vienna) SIRIGU Angela, Directrice de thèse, (DRCE, Université de Lyon, CNRS) 1 UNIVERSITE CLAUDE BERNARD - LYON 1 Président de l’Université M. -

Dopamine and Serotonin Metabolism in Parkinsonian Models
Dopamine and Serotonin Metabolism in Parkinsonian Models Carmen de la Fuente Barrigon UCL Great Ormond Street Institute of Child Health Thesis submitted for the degree of Doctor of Philosophy (PhD) awarded by University College London (UCL). Funded by Marie Skłodowska-Curie Actions of the European Union’s Seventh Framework Programme (FP7). APRIL 2018 1 I, Carmen de la Fuente Barrigon confirm that the work presented in this thesis is my own. Where information has been derived from other sources, I confirm that this has been indicated in the thesis. Signed ………………………………………….. Date ……………………………………………… 3 Dedico esta tesis a los pilares de mi vida. A mis padres, Tomás y Marisa. A mi hermana, Paloma. A mi amor, Francesco. Por vuestros sacrificios, paciencia, amor y apoyo incondicionales que me hacen ser quien soy hoy. 5 Abstract Parkinson’s disease (PD) is a neurodegenerative disorder caused by loss of dopaminergic neurons in the substantia nigra. Different pathogenic mechanisms have been implicated, including loss of mitochondrial complex I function and dysfunction of lysosomal glucocerebrosidase (GBA1) (Neumann et al., 2009; Schapira et al., 1990). Also, it has been hypothesised that serotonin metabolism could be affected in these patients due to the number of enzymes shared by both pathways (Albizu et al., 2011). This thesis considers the potential involvement of complex I and GBA1 in PD using HPLC analysis of changes in the extracellular levels of the metabolites of dopamine and serotonin, and the expression and activity of the enzymes of the dopamine pathway. Using SH-SY5Y cells, complex I deficiency was modelled using rotenone, and GBA1 deficiency was modelled using conduritol B epoxide (CBE). -

Neurophysiological and Neurohumoral Changes by High Frequency Stimulation (HFS) of Nucleus Accumbens (Nac)
From the Department of Neurosurgery of the University of Luebeck Director: Prof. Dr. med. Volker Tronnier Neurophysiological and neurohumoral changes by High Frequency Stimulation (HFS) of Nucleus Accumbens (NAc) Dissertation for Fulfillment of Requirements for the Doctoral Degree of the University of Luebeck from the Department of Natural Sciences Submitted by Ramya Varatharajan Thirugnanasambantham Born in Veeramanipatti-TN, India Luebeck, 2015 I Ramya Varatharajan Thirugnanasambantham Department of Neurosurgery University of Luebeck Ratzeburger allee 160 23562, Luebeck Germany First referee: Prof. Dr. Volker Tronnier Second referee: Prof. Dr. Thomas Martinetz Date of oral examination: 23.09.2015 Approved for printing, Luebeck: 30.09.2015 II Acknowledgements The hard work of so many people is involved in successful completion of this thesis, that a genuine list is virtually impossible. I convey my thanks and gratitude to Prof. Dr. med. Volker Tronnier, Department of Neurosurgery, for providing this opportunity to work under his guidance with this interesting topic. I express my deepest gratitude to Prof. Dr. med. Andreas Moser and Prof. Dr. rer. nat. Ulrich G. Hofmann for providing Laboratory Infrastructure, financial and moral support. Beyond scientific supervision, I was lucky to have the people from liquorlab, guiding me through all matters during my stay in Luebeck. I am thankful to Katharina Schnackenberg whose excellent experience in the laboratory and generous practical help was essential for the successful work at the lab. Thanks to Karin Wiegers, Marie Luis Reher and Gisa Brunk for being like friends and taking good care of me. I am very much thankful to Krishnakumar, Kevin Joseph, Sonya Neto, Mehrnaz Hazrati, Yijing Xie and Susanne Loeffler for their support, encouragement and timely suggestions. -

Imaging Elevated Brain Arachidonic Acid Signaling in Unanesthetized Serotonin Transporter (5-HTT)-Deficient Mice
Neuropsychopharmacology (2009) 34, 1695–1709 & 2009 Nature Publishing Group All rights reserved 0893-133X/09 $32.00 www.neuropsychopharmacology.org Imaging Elevated Brain Arachidonic Acid Signaling in Unanesthetized Serotonin Transporter (5-HTT)-Deficient Mice Mireille Basselin*,1, Meredith A Fox2, Lisa Chang1, Jane M Bell1, Dede Greenstein3, Mei Chen1, 2 1 Dennis L Murphy and Stanley I Rapoport 1Brain Physiology and Metabolism Section, National Institute on Aging, National Institutes of Health, Bethesda, MD, USA; 2Laboratory of Clinical Science, National Institutes of Health, Bethesda, MD, USA; 3Child Psychiatry Branch, National Institute of Mental Health, National Institutes of Health, Bethesda, MD, USA Certain polymorphisms reduce serotonin (5-HT) reuptake transporter (5-HTT) function and increase susceptibility to psychiatric +/À disorders. Heterozygous (5-HTT )-deficient mice, models for humans with these polymorphisms, have elevated brain 5-HT concentrations and behavioral abnormalities. As postsynaptic 5-HT2A/2C receptors are coupled to cytosolic phospholipase A2 (cPLA2), which releases arachidonic acid (AA) from membrane phospholipid, 5-HTT-deficient mice may have altered brain AA signaling and metabolism. To test this hypothesis, signaling was imaged as an AA incorporation coefficient k* in unanesthetized homozygous knockout À/À +/À +/+ (5-HTT ), 5-HTT and wild-type (5-HTT ), mice following saline (baseline) or 1.5 mg/kg s.c. DOI, a partial 5-HT2A/2C receptor agonist. Enzyme activities, metabolite concentrations, and head-twitch responses to DOI were also measured. Baseline k* was widely +/À À/À +/+ +/+ elevated by 20–70% in brains of 5-HTT and 5-HTT compared to 5-HTT mice. DOI increased k* in 5-HTT mice, but decreased k* in 5-HTT-deficient mice. -
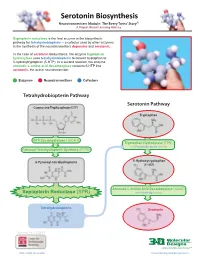
Serotonin Biosynthesis Neurotransmitters Module: the Beery Twins’ Story© a Project-Based Learning Activity
Serotonin Biosynthesis Neurotransmitters Module: The Beery Twins’ Story© A Project-Based Learning Activity Sepiapterin reductase is the final enzyme in the biosynthetic pathway for tetrahydrobiopterin – a cofactor used by other enzymes in the synthesis of the neurotransmitters dopamine and serotonin. In the case of serotonin biosynthesis, the enzyme tryptophan hydroxylase uses tetrahydrobiopterin to convert tryptophan to 5-hydroxytryptophan (5-HTP). In a second reaction, the enzyme aromatic L-amino acid decarboxylase converts 5-HTP into serotonin, the active neurotransmitter. Enzymes Neurotransmitters Cofactors Tetrahydrobiopterin Pathway Serotonin Pathway Guanosine Triphosphate (GTP) Tryptophan GTP Cyclohydrolase I (GCH1) Tryptophan Hydroxylase (TPH) with Tetrahydrobiopterin Cofactor Pyruvoyl-Tetrahydropterin Synthase (PTPS) 6-Pyruvoyl-tetrahydropterin 5-Hydroxytryptophan (5-HTP) Aromatic L-Amino Acid Decarboxylase (AAAD) Sepiapterin Reductase (SPR) with Vitamin B6 Cofactor Tetrahydrobiopterin Serotonin Version 1.4 -12/2015 ...where molecules become real TM http://cbm.msoe.edu www.3dmoleculardesigns.com Serotonin Biosynthesis Model Guide Neurotransmitters Module: The Beery Twins’ Story© A Project-Based Learning Activity TPH AAAD Tryptophan 5-Hydroxytryptophan Serotonin Tryptophan (Trp or W) is one of the 20 standard 5-Hydroxytryptophan, an intermediate The nal step in the serotonin biosynthesis amino acids and is an essential amino acid that molecule in the serotonin biosynthesis pathway requires the removal of the cannot be synthesized by the human body. pathway, is formed by the addition of a carboxylic acid group (COOH) from the Tryptophan is composed of the standard amino hydroxyl (OH) group to the fth carbon of the backbone of 5-hydroxytryptophan to form acid backbone with an indole ring side chain. indole ring of tryptophan. -

Modulation of Serotonin Signaling/Metabolism by Akkermansia Muciniphila and Its Extracellular Vesicles Through the Gut-Brain
www.nature.com/scientificreports OPEN Modulation of serotonin signaling/ metabolism by Akkermansia muciniphila and its extracellular vesicles through the gut‑brain axis in mice Rezvan Yaghoubfar1,2, Ava Behrouzi1,2, Fatemeh Ashrafan1,2, Arefeh Shahryari1,2, Hamid Reza Moradi3, Samira Choopani4, Shima Hadifar1,2, Farzam Vaziri1,2, Seyed Ali Nojoumi1,2, Abolfazl Fateh1,2*, Shohreh Khatami 5* & Seyed Davar Siadat1,2 Several studies have reported that the host‑microbe interactions in the gut modulate the host serotonin or 5‑hydroxytryptamine (5‑HT) system. Here, we evaluated the efects of Akkermansia muciniphila and its extracellular vesicles (EVs) on genes pertaining to the serotonergic system in the colon and hippocampus of mice. Male C57BL/6J mice were administered viable A. muciniphila and its EVs for 4 weeks. The serotonin levels in the colon, hippocampus, and serum of mice, as well as the human colon carcinoma cells (Caco‑2), were measured by ELISA assays. Also, the efects of A. muciniphila and its EVs on the expression of serotonin system genes in the colon and hippocampus were examined. A. muciniphila and its EVs may have a biological efect on the induction of serotonin levels in the colon and hippocampus of mice. Also, EVs increased the serotonin level in the Caco‑2 cell line. In contrast, both treatments decreased the serotonin level in the serum. Both the bacterium and its EVs had signifcant efects on the mRNA expression of genes, involved in serotonin signaling/ metabolism in the colon and hippocampus of mice. Moreover, A. muciniphila and its EVs afected the mRNA expression of infammatory cytokines (Il-10 and Tnf‑α) in the colon, however, there is no signifcant diference in infammatory cell infltrate in the histopathology of the colon. -

Serotonin Pathway in Cancer
International Journal of Molecular Sciences Review Serotonin Pathway in Cancer Pragathi Balakrishna 1, Sagila George 1, Hassan Hatoum 1,* and Sarbajit Mukherjee 2,* 1 Stephenson Cancer Center, University of Oklahoma Health Sciences Center, Oklahoma City, OK 73104, USA; [email protected] (P.B.); [email protected] (S.G.) 2 Roswell Park Comprehensive Cancer Center, Buffalo, NY 14263, USA * Correspondence: [email protected] (H.H.); [email protected] (S.M.) Abstract: Serotonin (5-hydroxytryptamine, 5-HT) is a biogenic monoamine produced from the essen- tial amino acid tryptophan. Serotonin’s role as a neurotransmitter in the central nervous system and a motility mediator in the gastrointestinal tract has been well defined, and its function in tumorigenesis in various cancers (gliomas, carcinoids, and carcinomas) is being studied. Many studies have shown a potential stimulatory effect of serotonin on cancer cell proliferation, invasion, dissemination, and tumor angiogenesis. Although the underlying mechanism is complex, it is proposed that serotonin levels in the tumor and its interaction with specific receptor subtypes are associated with disease progression. This review article describes serotonin’s role in cancer pathogenesis and the utility of the serotonin pathway as a potential therapeutic target in cancer treatment. Octreotide, an inhibitor of serotonin release, is used in well-differentiated neuroendocrine cancers, and the tryptophan hy- droxylase (TPH) inhibitor, telotristat, is currently being investigated in clinical trials to treat patients with metastatic neuroendocrine tumors and advanced cholangiocarcinoma. Several in vitro studies have shown the anticancer effect of 5-HT receptor antagonists in various cancers such as prostate cancer, breast cancer, urinary bladder, colorectal cancer, carcinoid, and small-cell lung cancer. -

Mechanism of Actions of Antidepressants: Beyond the Receptors
Derlemeler/Reviews Mechanism of Actions of Antidepressants: Beyond the Receptors Mechanism of Actions of Antidepressants: Beyond the Receptors Ayflegül Y›ld›z, M.D.1, Ali Saffet Gönül, M.D.2, Lut Tamam, M.D.3 ABSTRACT: MECHANISM OF ACTIONS OF ANTIDEPRESSANTS: BEYOND THE RECEPTORS Since the discovery of first antidepressants-monoamine oxidase inhibitors- a half century passed. There are now almost two- dozen antidepressant agents that work by nine distinct pharmacological mechanisms at the receptor level. However, opposite to the divergence in their pharmacological mechanisms at the receptor level, antidepressant drugs probably stimulate similar pathways in subcellular level. These subcellular events or so called beyond receptor effects are named neuroplasticity, and the mechanism may be called as adaptation. These after-receptor processes, through their effects on synaptic transmission, and gene expression are indeed capable of altering many molecular events in the brain. In this article, the mechanisms of actions of antidepressants at- and beyond- the receptors are discussed by documenting some of the evidence indicating such long-term alterations. Accordingly, the well-known effects of antidepressants on the receptor level are initiating events of antidepressant drug action, which enhance and prolong the actions of norepinephrine and/or serotonin and/or dopamine. Only if an adequate dose of an antidepressant is taken chronically, the increase in the synaptic norepinephrine and/or serotonin and/or dopamine stresses or perturbs the nervous -

Universidade Federal Do Ceará Faculdade De Medicina Programa De Pós-Graduação Em Farmacologia
Universidade Federal do Ceará Faculdade de Medicina Programa de Pós-graduação em Farmacologia Nayrton Flávio Moura Rocha Efeito sinérgico da associação entre L-triptofano e analgésicos não-opióides em modelos de nocicepção experimental em camundongos. Fortaleza 2013 NAYRTON FLÁVIO MOURA ROCHA Efeito sinérgico da associação entre L-triptofano e analgésicos não-opióides em modelos de nocicepção experimental em camundongos Tese submetida ao programa de pós- graduação em farmacologia da Universidade Federal do Ceará, como requisito parcial para obtenção do título de Doutor em Farmacologia. Orientação: Profa. Dra. Francisca Cléa Florenço de Sousa. Fortaleza 2013 Dados Internacionais de Catalogação na Publicação Universidade Federal do Ceará Biblioteca de Ciências da Saúde R574e Rocha, Nayrton Flávio Moura. Efeito sinérgico da associação entre L-triptofano e analgésicos não- opióides em modelos de nocicepção experimental em camundongos / Nayrton Flávio Moura Rocha. – 2013. 137 f. : il. Tese (Doutorado) - Universidade Federal do Ceará, Faculdade de Medicina, Programa de Pós-Graduação em Farmacologia, Fortaleza, 2013. Orientação: Profª Drª Francisca Cléa Florenço de Sousa 1. Serotonina. 2. Dipirona. 3. Acetaminofen. 4. Sinergismo Farmacológico. 5. Medição da Dor I. Título. CDD: 615.1 NAYRTON FLÁVIO MOURA ROCHA Efeito sinérgico da associação entre L-triptofano e analgésicos não-opióides em modelos de nocicepção experimental em camundongos. Tese submetida ao programa de pós- graduação em farmacologia da Universidade Federal do Ceará, como requisito parcial para obtenção do título de Doutor em Farmacologia. Aprovada em: / / . BANCA EXAMINADORA ___________________________________________________ Profa. Dra. Francisca Cléa Florenço de Sousa (Orientadora) Universidade Federal do Ceará (UFC) ___________________________________________________ Profa. Dra. Ana Maria Sampaio Assreuy Universidade Estadual do Ceará (UECE) ___________________________________________________ Prof. -

Serotonin Inhibition of Synaptic Transmission: G␣O Decreases the Abundance of UNC-13 at Release Sites
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Neuron, Vol. 24, 231±242, September, 1999, Copyright 1999 by Cell Press Serotonin Inhibition of Synaptic Transmission: Gao Decreases the Abundance of UNC-13 at Release Sites Stephen Nurrish,* Laurent Se galat,² including locomotion (Horvitz et al., 1982; Se galat et al., and Joshua M. Kaplan*³ 1995), egg laying (Trent et al., 1983; Waggoner et al., *Department of Molecular and Cell Biology 1998), and feeding (Avery and Horvitz, 1990). Six genes University of California, Berkeley are required for serotonin stimulation of the egg laying Berkeley, California 94720 muscles (Trent et al., 1983). Mutations in unc-2, which ² Institut de Pharmacologie encodes a voltage-dependent calcium channel subunit, Moleculare et Cellulaire prevent adaptation of animals to serotonin-induced inhi- CNRS bition of locomotion (Schafer and Kenyon, 1995). Muta- 660 Route des Lucioles tions that reduce the activity of goa-1 Gao confer partial Sophia Antipolis resistance to serotonin-induced inhibition of locomotion 06560 Valbonne (Mendel et al., 1995; Se galat et al., 1995). Taken together, France these studies define a set of genes that play some role in serotonin signal transduction. However, it remains unclear what cells are regulated by serotonin and how Summary serotonin regulates the activity of these cells. Serotonin often acts as a neuromodulator regulating We show that serotonin inhibits synaptic transmission transmission by other neurotransmitters. In some cases, at C. elegans neuromuscular junctions, and we de- serotonin has been shown to enhance synaptic trans- scribe a signaling pathway that mediates this effect. -

2012 NBDOA Middle School Notes
Guide to “The Neuroscience Behind Prescription and Illicit Drug Abuse” presentation Slide 1: Introduction Explain that you will be presenting information on how heroin, marijuana, prescription depressants, alcohol, cocaine, amphetamine and ecstasy alter the brain, and how these alterations cause mood and behavioral changes and eventually lead to addiction. Slide 2: Brain regions and neuronal pathways To understand how prescription drugs work, we need to first know a little neuroscience. Certain parts of the brain govern specific functions. Point to the sensory (blue), motor (orange) and visual cortex (yellow) areas to highlight their specific functions. Point to the cerebellum (pink) for coordination and to the hippocampus (green) for memory. Indicate that nerve cells or neurons connect one area to another via pathways to send and integrate information. The distances that neurons extend can be short or long. For example, point to the reward pathway (red). Explain that this pathway is activated when a person receives positive reinforcement for certain behaviors ("reward"). Indicate that you will explain how this happens when a person takes an addictive drug. As another example, point to the thalamus (magenta). This structure receives information about pain coming from the body (magenta line within the spinal cord), and passes the information up to the cortex. The thalamus is also involved in drug dependence. Tell the audience that you can look at the neuronal connections in more detail. Slide 3: Neuronal structure Remind the audience that pathways are made up of neurons like these (viewed through a high-powered microscope). Explain the normal direction of impulse flow. Point to the cell on the top and indicate that electrical impulses (the action potential) flow in the direction toward the terminal. -

Molecular Neurobiology and Promising New Treatment in Depression
International Journal of Molecular Sciences Article Molecular Neurobiology and Promising New Treatment in Depression Sang Won Jeon and Yong-Ku Kim * Department of Psychiatry, College of Medicine, Korea University, Ansan Hospital, 123, Jeokgeum-ro, Danwon-gu, Ansan-si, Gyeonggi-do, Seoul 15355, Korea; [email protected] * Correspondence: [email protected]; Tel.: +82-31-412-5140; Fax: +82-31-412-5144 Academic Editor: Kenji Hashimoto Received: 26 January 2016; Accepted: 7 March 2016; Published: 15 March 2016 Abstract: The limited effects of currently available antidepressants are becoming an urgent issue in depression research. It takes a long time to determine treatment effects, and the overall remission rate is low. Although we expect the development of non-monoamine antidepressants in the near future, efforts in this regard over the past several decades have not yet been compensated. Thus, researchers and clinicians should clarify the neurobiological mechanisms of integrated modulators that regulate changes in genes, cells, the brain, and behaviors associated with depression. In this study, we review molecular neurobiological theories and new treatments for depression. Beyond neuroanatomy and monoamine theory, we discuss cells and molecules, neural plasticity, neurotrophisms, endocrine mechanisms, immunological mechanisms, genetics, circadian rhythms, and metabolic regulation in depression. In addition, we introduce the possibility of new antidepressant drug development using protein translation signaling (mTOR) pathways. Keywords: depression; neurobiology; antidepressant; neural plasticity; BDNF; endocrine; immune; gene; mTOR 1. Introduction Depression is currently one of the most important causes of mortality and morbidity, which occurs in all genders, ages, and social backgrounds. Serious problems, such as suicidal behavior, is frequently occurring in the patients with depression.