The Placenta
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Manual Annotation and Analysis of the Defensin Gene Cluster in the C57BL
BMC Genomics BioMed Central Research article Open Access Manual annotation and analysis of the defensin gene cluster in the C57BL/6J mouse reference genome Clara Amid*†1, Linda M Rehaume*†2, Kelly L Brown2,3, James GR Gilbert1, Gordon Dougan1, Robert EW Hancock2 and Jennifer L Harrow1 Address: 1Wellcome Trust Sanger Institute, Wellcome Trust Genome Campus, Hinxton, Cambridgeshire CB10 1SA, UK, 2University of British Columbia, Centre for Microbial Disease & Immunity Research, 2259 Lower Mall, Vancouver, BC, V6T 1Z4, Canada and 3Department of Rheumatology and Inflammation Research, Göteborg University, Guldhedsgatan 10, S-413 46 Göteborg, Sweden Email: Clara Amid* - [email protected]; Linda M Rehaume* - [email protected]; Kelly L Brown - [email protected]; James GR Gilbert - [email protected]; Gordon Dougan - [email protected]; Robert EW Hancock - [email protected]; Jennifer L Harrow - [email protected] * Corresponding authors †Equal contributors Published: 15 December 2009 Received: 15 May 2009 Accepted: 15 December 2009 BMC Genomics 2009, 10:606 doi:10.1186/1471-2164-10-606 This article is available from: http://www.biomedcentral.com/1471-2164/10/606 © 2009 Amid et al; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Abstract Background: Host defense peptides are a critical component of the innate immune system. Human alpha- and beta-defensin genes are subject to copy number variation (CNV) and historically the organization of mouse alpha-defensin genes has been poorly defined. -
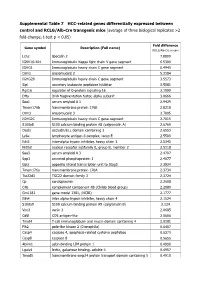
Supplemental Table 7 HCC-Related Genes Differentially Expressed Between Control and RCLG/Alb-Cre Transgenic Mice
Supplemental Table 7 HCC-related genes differentially expressed between control and RCLG/Alb-Cre transgenic mice (average of three biological replicates >2 fold-change, t-test p < 0.05) Fold difference Gene symbol Description (Full name) (RCLG/Alb-Cre vs con) Lcn2 lipocalin 2 7.8899 IGKV16-104 Immunoglobulin Kappa light chain V gene segment 6.5300 IGHG1 Immunoglobulin heavy chain C gene segment 6.4945 Orm2 orosomucoid 2 5.3184 IGHG2B Immunoglobulin heavy chain C gene segment 3.5573 Slpi secretory leukocyte peptidase inhibitor 3.5081 Rgs16 regulator of G-protein signaling 16 3.1999 Dffa DNA fragmentation factor, alpha subunit 3.0666 Saa1 serum amyloid A 1 2.9429 Tmem176b transmembrane protein 176B 2.8218 Orm3 orosomucoid 3 2.7805 IGHG2C Immunoglobulin heavy chain C gene segment 2.7015 S100a8 S100 calcium binding protein A8 (calgranulin A) 2.6769 Ocel1 occludin/ELL domain containing 1 2.6553 Ly6e lymphocyte antigen 6 complex, locus E 2.5509 Itih3 inter-alpha trypsin inhibitor, heavy chain 3 2.5345 Nr0b2 nuclear receptor subfamily 0, group B, member 2 2.5118 Saa3 serum amyloid A 3 2.4707 Spp1 secreted phosphoprotein 1 2.4077 Gats opposite strand transcription unit to Stag3 2.3934 Tmem176a transmembrane protein 176A 2.3734 Tsc22d3 TSC22 domain family 3 2.3724 Cp ceruloplasmin 2.2608 C4b complement component 4B (Childo blood group) 2.2089 Gm1381 gene model 1381, (NCBI) 2.1777 Itih4 inter alpha-trypsin inhibitor, heavy chain 4 2.1524 S100a9 S100 calcium binding protein A9 (calgranulin B) 2.124 Vnn3 vanin 3 2.0685 Cd5l CD5 antigen-like 2.0606 -

Mapping DNA Structural Variation in Dogs
Downloaded from genome.cshlp.org on October 3, 2021 - Published by Cold Spring Harbor Laboratory Press Resource Mapping DNA structural variation in dogs Wei-Kang Chen,1,4 Joshua D. Swartz,1,4,5 Laura J. Rush,2 and Carlos E. Alvarez1,3,6 1Center for Molecular and Human Genetics, The Research Institute at Nationwide Children’s Hospital, Columbus, Ohio 43205, USA; 2Department of Veterinary Biosciences, The Ohio State University, Columbus, Ohio 43210, USA; 3Department of Pediatrics, The Ohio State University College of Medicine, Columbus, Ohio 43210, USA DNA structural variation (SV) comprises a major portion of genetic diversity, but its biological impact is unclear. We propose that the genetic history and extraordinary phenotypic variation of dogs make them an ideal mammal in which to study the effects of SV on biology and disease. The hundreds of existing dog breeds were created by selection of extreme morphological and behavioral traits. And along with those traits, each breed carries increased risk for different diseases. We used array CGH to create the first map of DNA copy number variation (CNV) or SV in dogs. The extent of this variation, and some of the gene classes affected, are similar to those of mice and humans. Most canine CNVs affect genes, including disease and candidate disease genes, and are thus likely to be functional. We identified many CNVs that may be breed or breed class specific. Cluster analysis of CNV regions showed that dog breeds tend to group according to breed classes. Our combined findings suggest many CNVs are (1) in linkage disequilibrium with flanking sequence, and (2) associated with breed-specific traits. -

4-6 Weeks Old Female C57BL/6 Mice Obtained from Jackson Labs Were Used for Cell Isolation
Methods Mice: 4-6 weeks old female C57BL/6 mice obtained from Jackson labs were used for cell isolation. Female Foxp3-IRES-GFP reporter mice (1), backcrossed to B6/C57 background for 10 generations, were used for the isolation of naïve CD4 and naïve CD8 cells for the RNAseq experiments. The mice were housed in pathogen-free animal facility in the La Jolla Institute for Allergy and Immunology and were used according to protocols approved by the Institutional Animal Care and use Committee. Preparation of cells: Subsets of thymocytes were isolated by cell sorting as previously described (2), after cell surface staining using CD4 (GK1.5), CD8 (53-6.7), CD3ε (145- 2C11), CD24 (M1/69) (all from Biolegend). DP cells: CD4+CD8 int/hi; CD4 SP cells: CD4CD3 hi, CD24 int/lo; CD8 SP cells: CD8 int/hi CD4 CD3 hi, CD24 int/lo (Fig S2). Peripheral subsets were isolated after pooling spleen and lymph nodes. T cells were enriched by negative isolation using Dynabeads (Dynabeads untouched mouse T cells, 11413D, Invitrogen). After surface staining for CD4 (GK1.5), CD8 (53-6.7), CD62L (MEL-14), CD25 (PC61) and CD44 (IM7), naïve CD4+CD62L hiCD25-CD44lo and naïve CD8+CD62L hiCD25-CD44lo were obtained by sorting (BD FACS Aria). Additionally, for the RNAseq experiments, CD4 and CD8 naïve cells were isolated by sorting T cells from the Foxp3- IRES-GFP mice: CD4+CD62LhiCD25–CD44lo GFP(FOXP3)– and CD8+CD62LhiCD25– CD44lo GFP(FOXP3)– (antibodies were from Biolegend). In some cases, naïve CD4 cells were cultured in vitro under Th1 or Th2 polarizing conditions (3, 4). -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

SLURP1 Gene Secreted LY6/PLAUR Domain Containing 1
SLURP1 gene secreted LY6/PLAUR domain containing 1 Normal Function The SLURP1 gene provides instructions for making a protein called secreted Ly6/uPAR- related protein-1 (SLURP-1). This protein is found in skin cells and other cells that line the surfaces and cavities of the body. Like other Ly6/uPAR-related proteins, SLURP-1 folds into a particular shape and is thought to attach (bind) to other proteins called receptors to carry out signaling within cells. However, SLURP-1's role in the skin and the rest of the body is not completely understood. Laboratory studies show that SLURP-1 can bind to nicotinic acetylcholine receptors ( nAChRs). SLURP-1 specifically interacts with the alpha7 (a 7) subunit, which is a piece of some nAChRs. Nicotinic acetylcholine receptors are best known for their role in chemical signaling between nerve cells, but they are also found in other tissues. In the skin, nAChRs regulate the activity of genes involved in the growth and division ( proliferation), maturation (differentiation), and survival of cells. Through its interaction with these receptors, SLURP-1 may be involved in skin growth and development. Health Conditions Related to Genetic Changes Mal de Meleda At least 15 mutations in the SLURP1 gene have been found to cause mal de Meleda, a rare disorder characterized by tough, thickened skin on the hands and feet. On the palms and soles, the thickening is known as palmoplantar keratoderma; the thickened skin also extends to the backs of the hands and feet and up to the wrists and ankles. The SLURP1 gene mutations involved in this condition lead to production of an altered SLURP-1 protein that is unstable and quickly broken down, if any protein is produced at all. -

Hippo and Sonic Hedgehog Signalling Pathway Modulation of Human Urothelial Tissue Homeostasis
Hippo and Sonic Hedgehog signalling pathway modulation of human urothelial tissue homeostasis Thomas Crighton PhD University of York Department of Biology November 2020 Abstract The urinary tract is lined by a barrier-forming, mitotically-quiescent urothelium, which retains the ability to regenerate following injury. Regulation of tissue homeostasis by Hippo and Sonic Hedgehog signalling has previously been implicated in various mammalian epithelia, but limited evidence exists as to their role in adult human urothelial physiology. Focussing on the Hippo pathway, the aims of this thesis were to characterise expression of said pathways in urothelium, determine what role the pathways have in regulating urothelial phenotype, and investigate whether the pathways are implicated in muscle-invasive bladder cancer (MIBC). These aims were assessed using a cell culture paradigm of Normal Human Urothelial (NHU) cells that can be manipulated in vitro to represent different differentiated phenotypes, alongside MIBC cell lines and The Cancer Genome Atlas resource. Transcriptomic analysis of NHU cells identified a significant induction of VGLL1, a poorly understood regulator of Hippo signalling, in differentiated cells. Activation of upstream transcription factors PPARγ and GATA3 and/or blockade of active EGFR/RAS/RAF/MEK/ERK signalling were identified as mechanisms which induce VGLL1 expression in NHU cells. Ectopic overexpression of VGLL1 in undifferentiated NHU cells and MIBC cell line T24 resulted in significantly reduced proliferation. Conversely, knockdown of VGLL1 in differentiated NHU cells significantly reduced barrier tightness in an unwounded state, while inhibiting regeneration and increasing cell cycle activation in scratch-wounded cultures. A signalling pathway previously observed to be inhibited by VGLL1 function, YAP/TAZ, was unaffected by VGLL1 manipulation. -

Organization, Evolution and Functions of the Human and Mouse Ly6/Upar Family Genes Chelsea L
Loughner et al. Human Genomics (2016) 10:10 DOI 10.1186/s40246-016-0074-2 GENE FAMILY UPDATE Open Access Organization, evolution and functions of the human and mouse Ly6/uPAR family genes Chelsea L. Loughner1, Elspeth A. Bruford2, Monica S. McAndrews3, Emili E. Delp1, Sudha Swamynathan1 and Shivalingappa K. Swamynathan1,4,5,6,7* Abstract Members of the lymphocyte antigen-6 (Ly6)/urokinase-type plasminogen activator receptor (uPAR) superfamily of proteins are cysteine-rich proteins characterized by a distinct disulfide bridge pattern that creates the three-finger Ly6/uPAR (LU) domain. Although the Ly6/uPAR family proteins share a common structure, their expression patterns and functions vary. To date, 35 human and 61 mouse Ly6/uPAR family members have been identified. Based on their subcellular localization, these proteins are further classified as GPI-anchored on the cell membrane, or secreted. The genes encoding Ly6/uPAR family proteins are conserved across different species and are clustered in syntenic regions on human chromosomes 8, 19, 6 and 11, and mouse Chromosomes 15, 7, 17, and 9, respectively. Here, we review the human and mouse Ly6/uPAR family gene and protein structure and genomic organization, expression, functions, and evolution, and introduce new names for novel family members. Keywords: Ly6/uPAR family, LU domain, Three-finger domain, uPAR, Lymphocytes, Neutrophils Introduction an overview of the Ly6/uPAR gene family and their gen- The lymphocyte antigen-6 (Ly6)/urokinase-type plas- omic organization, evolution, as well as functions, and minogen activator receptor (uPAR) superfamily of struc- provide a nomenclature system for the newly identified turally related proteins is characterized by the LU members of this family. -

The Roles of RNA Polymerase I and III Subunits Polr1a, Polr1c, and Polr1d in Craniofacial Development BY
The roles of RNA Polymerase I and III subunits Polr1a, Polr1c, and Polr1d in craniofacial development BY © 2016 Kristin Emily Noack Watt Submitted to the graduate degree program in Anatomy and Cell Biology and to the Graduate Faculty of The University of Kansas Medical Center in partial fulfillment of the requirements for the degree of Doctor of Philosophy. Paul Trainor, Co-Chairperson Brenda Rongish, Co-Chairperson Brian Andrews Jennifer Gerton Tatjana Piotrowski Russell Swerdlow Date Defended: January 26, 2016 The Dissertation Committee for Kristin Watt certifies that this is the approved version of the following dissertation: The roles of RNA Polymerase I and III subunits Polr1a, Polr1c, and Polr1d in craniofacial development Paul Trainor, Co-Chairperson Brenda Rongish, Co-Chairperson Date approved: February 2, 2016 ii Abstract Craniofacial anomalies account for approximately one-third of all birth defects. Two examples of syndromes associated with craniofacial malformations are Treacher Collins syndrome and Acrofacial Dysostosis, Cincinnati type which have phenotypic overlap including deformities of the eyes, ears, and facial bones. Mutations in TCOF1, POLR1C or POLR1D may cause Treacher Collins syndrome while mutations in POLR1A may cause Acrofacial Dysostosis, Cincinnati type. TCOF1 encodes the nucleolar phosphoprotein Treacle, which functions in rRNA transcription and modification. Previous studies demonstrated that Tcof1 mutations in mice result in reduced ribosome biogenesis and increased neuroepithelial apoptosis. This diminishes the neural crest cell (NCC) progenitor population which contribute to the development of the cranial skeleton. In contrast, apart from being subunits of RNA Polymerases (RNAP) I and/or III, nothing is known about the function of POLR1A, POLR1C, and POLR1D during embryonic and craniofacial development. -

MOCHI Enables Discovery of Heterogeneous Interactome Modules in 3D Nucleome
Downloaded from genome.cshlp.org on October 4, 2021 - Published by Cold Spring Harbor Laboratory Press MOCHI enables discovery of heterogeneous interactome modules in 3D nucleome Dechao Tian1,# , Ruochi Zhang1,# , Yang Zhang1, Xiaopeng Zhu1, and Jian Ma1,* 1Computational Biology Department, School of Computer Science, Carnegie Mellon University, Pittsburgh, PA 15213, USA #These two authors contributed equally *Correspondence: [email protected] Contact To whom correspondence should be addressed: Jian Ma School of Computer Science Carnegie Mellon University 7705 Gates-Hillman Complex 5000 Forbes Avenue Pittsburgh, PA 15213 Phone: +1 (412) 268-2776 Email: [email protected] 1 Downloaded from genome.cshlp.org on October 4, 2021 - Published by Cold Spring Harbor Laboratory Press Abstract The composition of the cell nucleus is highly heterogeneous, with different constituents forming complex interactomes. However, the global patterns of these interwoven heterogeneous interactomes remain poorly understood. Here we focus on two different interactomes, chromatin interaction network and gene regulatory network, as a proof-of-principle, to identify heterogeneous interactome modules (HIMs), each of which represents a cluster of gene loci that are in spatial contact more frequently than expected and that are regulated by the same group of transcription factors. HIM integrates transcription factor binding and 3D genome structure to reflect “transcriptional niche” in the nucleus. We develop a new algorithm MOCHI to facilitate the discovery of HIMs based on network motif clustering in heterogeneous interactomes. By applying MOCHI to five different cell types, we found that HIMs have strong spatial preference within the nucleus and exhibit distinct functional properties. Through integrative analysis, this work demonstrates the utility of MOCHI to identify HIMs, which may provide new perspectives on the interplay between transcriptional regulation and 3D genome organization. -

Detailed Characterization of Human Induced Pluripotent Stem Cells Manufactured for Therapeutic Applications
Stem Cell Rev and Rep DOI 10.1007/s12015-016-9662-8 Detailed Characterization of Human Induced Pluripotent Stem Cells Manufactured for Therapeutic Applications Behnam Ahmadian Baghbaderani 1 & Adhikarla Syama2 & Renuka Sivapatham3 & Ying Pei4 & Odity Mukherjee2 & Thomas Fellner1 & Xianmin Zeng3,4 & Mahendra S. Rao5,6 # The Author(s) 2016. This article is published with open access at Springerlink.com Abstract We have recently described manufacturing of hu- help determine which set of tests will be most useful in mon- man induced pluripotent stem cells (iPSC) master cell banks itoring the cells and establishing criteria for discarding a line. (MCB) generated by a clinically compliant process using cord blood as a starting material (Baghbaderani et al. in Stem Cell Keywords Induced pluripotent stem cells . Embryonic stem Reports, 5(4), 647–659, 2015). In this manuscript, we de- cells . Manufacturing . cGMP . Consent . Markers scribe the detailed characterization of the two iPSC clones generated using this process, including whole genome se- quencing (WGS), microarray, and comparative genomic hy- Introduction bridization (aCGH) single nucleotide polymorphism (SNP) analysis. We compare their profiles with a proposed calibra- Induced pluripotent stem cells (iPSCs) are akin to embryonic tion material and with a reporter subclone and lines made by a stem cells (ESC) [2] in their developmental potential, but dif- similar process from different donors. We believe that iPSCs fer from ESC in the starting cell used and the requirement of a are likely to be used to make multiple clinical products. We set of proteins to induce pluripotency [3]. Although function- further believe that the lines used as input material will be used ally identical, iPSCs may differ from ESC in subtle ways, at different sites and, given their immortal status, will be used including in their epigenetic profile, exposure to the environ- for many years or even decades. -

Human Induced Pluripotent Stem Cell–Derived Podocytes Mature Into Vascularized Glomeruli Upon Experimental Transplantation
BASIC RESEARCH www.jasn.org Human Induced Pluripotent Stem Cell–Derived Podocytes Mature into Vascularized Glomeruli upon Experimental Transplantation † Sazia Sharmin,* Atsuhiro Taguchi,* Yusuke Kaku,* Yasuhiro Yoshimura,* Tomoko Ohmori,* ‡ † ‡ Tetsushi Sakuma, Masashi Mukoyama, Takashi Yamamoto, Hidetake Kurihara,§ and | Ryuichi Nishinakamura* *Department of Kidney Development, Institute of Molecular Embryology and Genetics, and †Department of Nephrology, Faculty of Life Sciences, Kumamoto University, Kumamoto, Japan; ‡Department of Mathematical and Life Sciences, Graduate School of Science, Hiroshima University, Hiroshima, Japan; §Division of Anatomy, Juntendo University School of Medicine, Tokyo, Japan; and |Japan Science and Technology Agency, CREST, Kumamoto, Japan ABSTRACT Glomerular podocytes express proteins, such as nephrin, that constitute the slit diaphragm, thereby contributing to the filtration process in the kidney. Glomerular development has been analyzed mainly in mice, whereas analysis of human kidney development has been minimal because of limited access to embryonic kidneys. We previously reported the induction of three-dimensional primordial glomeruli from human induced pluripotent stem (iPS) cells. Here, using transcription activator–like effector nuclease-mediated homologous recombination, we generated human iPS cell lines that express green fluorescent protein (GFP) in the NPHS1 locus, which encodes nephrin, and we show that GFP expression facilitated accurate visualization of nephrin-positive podocyte formation in