Whole Genome Microarray Analysis of Gene Expression in Subjects with Fragile X Syndrome Douglas C
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Role of Molecular Scaffolds at the Active Zone in Synaptic Vesicle Distribution and Release Probability
The Role of Molecular Scaffolds at the Active Zone in Synaptic Vesicle Distribution and Release Probability DISSERTATION To obtain the academic degree Doctor rerum naturalium (Dr. rer. nat.) submitted to the Department of Biology, Chemistry and Pharmacy Freie Universität Berlin Berlin, 2016 by Christina Beis (née Hollmann) This thesis was completed under the supervision of Prof. Dr. Stephan Sigrist from February 2011 to June 2016 at the Institute for Biology/ Genetics of Freie Universität Berlin, Germany. 1st reviewer: Prof. Dr. Stephan Sigrist 2nd reviewer: Prof. Dr. Hans-Joachim Pflüger Date of Defense: 21.11.2016 Statement of Authorship I hereby declare that the work presented in this thesis has been written independently and without inappropriate support. All sources of information are referenced. I hereby declare that this thesis has not been submitted, either in the same or in a different form, to this or any other university for a degree. ____________________________ Christina Beis Contents 1. Summary / Zusammenfassung .................................................................................................. 1 Summary....................................................................................................................................... 1 Zusammenfassung ........................................................................................................................ 3 2. Introduction .................................................................................................................................... -

Primate Specific Retrotransposons, Svas, in the Evolution of Networks That Alter Brain Function
Title: Primate specific retrotransposons, SVAs, in the evolution of networks that alter brain function. Olga Vasieva1*, Sultan Cetiner1, Abigail Savage2, Gerald G. Schumann3, Vivien J Bubb2, John P Quinn2*, 1 Institute of Integrative Biology, University of Liverpool, Liverpool, L69 7ZB, U.K 2 Department of Molecular and Clinical Pharmacology, Institute of Translational Medicine, The University of Liverpool, Liverpool L69 3BX, UK 3 Division of Medical Biotechnology, Paul-Ehrlich-Institut, Langen, D-63225 Germany *. Corresponding author Olga Vasieva: Institute of Integrative Biology, Department of Comparative genomics, University of Liverpool, Liverpool, L69 7ZB, [email protected] ; Tel: (+44) 151 795 4456; FAX:(+44) 151 795 4406 John Quinn: Department of Molecular and Clinical Pharmacology, Institute of Translational Medicine, The University of Liverpool, Liverpool L69 3BX, UK, [email protected]; Tel: (+44) 151 794 5498. Key words: SVA, trans-mobilisation, behaviour, brain, evolution, psychiatric disorders 1 Abstract The hominid-specific non-LTR retrotransposon termed SINE–VNTR–Alu (SVA) is the youngest of the transposable elements in the human genome. The propagation of the most ancient SVA type A took place about 13.5 Myrs ago, and the youngest SVA types appeared in the human genome after the chimpanzee divergence. Functional enrichment analysis of genes associated with SVA insertions demonstrated their strong link to multiple ontological categories attributed to brain function and the disorders. SVA types that expanded their presence in the human genome at different stages of hominoid life history were also associated with progressively evolving behavioural features that indicated a potential impact of SVA propagation on a cognitive ability of a modern human. -

Development of a Human Mitochondrial Oligonucleotide
BMC Genomics BioMed Central Research article Open Access Development of a human mitochondrial oligonucleotide microarray (h-MitoArray) and gene expression analysis of fibroblast cell lines from 13 patients with isolated F1Fo ATP synthase deficiency Alena Жížková1,2,5, Viktor Stránecký1,2, Robert Ivánek1,2,4, Hana Hartmannová1,2, Lenka Nosková2, Lenka Piherová1,2, Markéta Tesařová1,3, Hana Hansíková1,3, Tomáš Honzík3, JiříZeman1,3, Petr Divina4, Andrea Potocká1,5, Jan Paul1,5, Wolfgang Sperl6, Johannes A Mayr6, Sara Seneca7, Josef Houštĕk1,5 and Stanislav Kmoch*1,2 Address: 1Center for Applied Genomics, 1st Faculty of Medicine, Charles University, Prague, Czech Republic, 2Institute of Inherited Metabolic Disorders, 1st Faculty of Medicine, Charles University, Prague, Czech Republic, 3Department of Pediatrics, 1st Faculty of Medicine, Charles University, Prague, Czech Republic, 4Institute of Molecular Genetics, Academy of Science of the Czech Republic, Prague, Czech Republic, 5Department of Bioenergetics, Institute of Physiology, Academy of Science of the Czech Republic, Prague, Czech Republic, 6Department of Pediatrics, Paracelsus Medical University, Salzburg, Austria and 7Center of Medical Genetics, Free University Brussels, Brussels, Belgium Email: Alena Жížková - [email protected]; Viktor Stránecký - [email protected]; Robert Ivánek - [email protected]; Hana Hartmannová - [email protected]; Lenka Nosková - [email protected]; Lenka Piherová - [email protected]; Markéta Tesařová - [email protected]; Hana Hansíková - -

High-Fidelity CRISPR/Cas9
ARTICLE DOI: 10.1038/s41467-018-05766-5 OPEN High-fidelity CRISPR/Cas9- based gene-specific hydroxymethylation rescues gene expression and attenuates renal fibrosis Xingbo Xu1,2, Xiaoying Tan2,3, Björn Tampe 3, Tim Wilhelmi1,2, Melanie S. Hulshoff1,2,4, Shoji Saito3, Tobias Moser 5, Raghu Kalluri6, Gerd Hasenfuss1,2, Elisabeth M. Zeisberg1,2 & Michael Zeisberg2,3 fi 1234567890():,; While suppression of speci c genes through aberrant promoter methylation contributes to different diseases including organ fibrosis, gene-specific reactivation technology is not yet available for therapy. TET enzymes catalyze hydroxymethylation of methylated DNA, reac- tivating gene expression. We here report generation of a high-fidelity CRISPR/Cas9-based gene-specific dioxygenase by fusing an endonuclease deactivated high-fidelity Cas9 (dHFCas9) to TET3 catalytic domain (TET3CD), targeted to specific genes by guiding RNAs (sgRNA). We demonstrate use of this technology in four different anti-fibrotic genes in different cell types in vitro, among them RASAL1 and Klotho, both hypermethylated in kidney fibrosis. Furthermore, in vivo lentiviral delivery of the Rasal1-targeted fusion protein to interstitial cells and of the Klotho-targeted fusion protein to tubular epithelial cells each results in specific gene reactivation and attenuation of fibrosis, providing gene-specific demethylating technology in a disease model. 1 Department of Cardiology and Pneumology, University Medical Center Göttingen, Robert-Koch-Str. 40, 37075 Göttingen, Germany. 2 German Center for Cardiovascular Research (DZHK) Partner Site, Göttingen, Germany. 3 Department of Nephrology and Rheumatology, University Medical Center Göttingen, Robert-Koch-Str. 40, 37075 Göttingen, Germany. 4 Department of Pathology and Medical Biology, University Medical Center Groningen, Hanzeplein 1, 9713 Groningen, GZ, Netherlands. -

Role of Amylase in Ovarian Cancer Mai Mohamed University of South Florida, [email protected]
University of South Florida Scholar Commons Graduate Theses and Dissertations Graduate School July 2017 Role of Amylase in Ovarian Cancer Mai Mohamed University of South Florida, [email protected] Follow this and additional works at: http://scholarcommons.usf.edu/etd Part of the Pathology Commons Scholar Commons Citation Mohamed, Mai, "Role of Amylase in Ovarian Cancer" (2017). Graduate Theses and Dissertations. http://scholarcommons.usf.edu/etd/6907 This Dissertation is brought to you for free and open access by the Graduate School at Scholar Commons. It has been accepted for inclusion in Graduate Theses and Dissertations by an authorized administrator of Scholar Commons. For more information, please contact [email protected]. Role of Amylase in Ovarian Cancer by Mai Mohamed A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy Department of Pathology and Cell Biology Morsani College of Medicine University of South Florida Major Professor: Patricia Kruk, Ph.D. Paula C. Bickford, Ph.D. Meera Nanjundan, Ph.D. Marzenna Wiranowska, Ph.D. Lauri Wright, Ph.D. Date of Approval: June 29, 2017 Keywords: ovarian cancer, amylase, computational analyses, glycocalyx, cellular invasion Copyright © 2017, Mai Mohamed Dedication This dissertation is dedicated to my parents, Ahmed and Fatma, who have always stressed the importance of education, and, throughout my education, have been my strongest source of encouragement and support. They always believed in me and I am eternally grateful to them. I would also like to thank my brothers, Mohamed and Hussien, and my sister, Mariam. I would also like to thank my husband, Ahmed. -

Staufen 1 Does Not Play a Role in NPC Asymmetric Divisions but Regulates Cellular Positioning During Corticogenesis
Staufen 1 does not play a role in NPC asymmetric divisions but regulates cellular positioning during corticogenesis by Christopher Kuc A Thesis presented to The University of Guelph In partial fulfilment of requirements for the degree of Master of Science in Molecular and Cellular Biology Guelph, Ontario, Canada © Christopher Kuc, September 2018 ABSTRACT INVESTIGATING THE ROLE OF STAUFEN1 IN ASYMMETRIC NEURAL PRECURSOR CELL DIVISIONS IN THE DEVELOPING CEREBRAL CORTEX Christopher Kuc Advisors: Dr. John Vessey University of Guelph, 2018 Cerebral cortex development relies on asymmetric divisions of neural precursor cells (NPCs) to produce a recurring NPC and a differentiated neuron. Asymmetric divisions are promoted by the differential localization of cell fate determinants between daughter cells. Staufen 1 (Stau1) is an RNA-binding protein known to localize mRNA in mature hippocampal neurons. However, its expression pattern and role in the developing mammalian cortex remains unknown. In this study, Stau1 mRNA and protein were found to be expressed in all cells examined and was temporally and spatially characterized across development. Upon shRNA-mediated knockdown of Stau1 in primary cortical cultures, NPCs retained the ability to self-renew and generate neurons despite the loss of Stau1 expression. This said, in vivo knockdown of Stau1 demonstrated that it may play a role in anchoring NPCs to the ventricular zone during cortical development. ACKNOWLEDGMENTS I would first like to thank my advisor Dr. John Vessey. Throughout these 2 years, you have provided me with an invaluable opportunity and played an instrumental role in shaping me as a scientist. The guidance, support and expertise you have provided me will be always appreciated and never forgotten. -

Pharmacological Mechanisms Underlying the Hepatoprotective Effects of Ecliptae Herba on Hepatocellular Carcinoma
Hindawi Evidence-Based Complementary and Alternative Medicine Volume 2021, Article ID 5591402, 17 pages https://doi.org/10.1155/2021/5591402 Research Article Pharmacological Mechanisms Underlying the Hepatoprotective Effects of Ecliptae herba on Hepatocellular Carcinoma Botao Pan ,1 Wenxiu Pan ,2 Zheng Lu ,3 and Chenglai Xia 1,4 1Affiliated Foshan Maternity & Child Healthcare Hospital, Southern Medical University, Foshan 528000, China 2Department of Laboratory, Fifth People’s Hospital of Foshan, Foshan 528000, China 3Wuzhou Maternal and Child Health-Care Hospital, Wuzhou 543000, China 4School of Pharmaceutical Sciences, Southern Medical University, Guangzhou 510515, China Correspondence should be addressed to Zheng Lu; [email protected] and Chenglai Xia; [email protected] Received 30 January 2021; Revised 31 May 2021; Accepted 3 July 2021; Published 16 July 2021 Academic Editor: Hongcai Shang Copyright © 2021 Botao Pan et al. -is is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Background. -e number of hepatocellular carcinoma (HCC) cases worldwide has increased significantly. As a traditional Chinese medicine (TCM) with a long history, Ecliptae herba (EH) has been widely used in HCC patients in China, but its hepatoprotective mechanism is still unclear. Methods. In this study, we applied a network pharmacology-based strategy and experimental ver- ification to systematically unravel the underlying mechanisms of EH against HCC. First, six active ingredients of EH were screened from the Traditional Chinese Medicine Systems Pharmacology Database and Analysis Platform (TCMSP) by the ADME method. Subsequently, 52 potential targets of 6 active ingredients acting on HCC were screened from various databases, including TCMSP, DGIdb, SwissTargetPrediction, CTD, and GeneCards. -

Analysis of the Indacaterol-Regulated Transcriptome in Human Airway
Supplemental material to this article can be found at: http://jpet.aspetjournals.org/content/suppl/2018/04/13/jpet.118.249292.DC1 1521-0103/366/1/220–236$35.00 https://doi.org/10.1124/jpet.118.249292 THE JOURNAL OF PHARMACOLOGY AND EXPERIMENTAL THERAPEUTICS J Pharmacol Exp Ther 366:220–236, July 2018 Copyright ª 2018 by The American Society for Pharmacology and Experimental Therapeutics Analysis of the Indacaterol-Regulated Transcriptome in Human Airway Epithelial Cells Implicates Gene Expression Changes in the s Adverse and Therapeutic Effects of b2-Adrenoceptor Agonists Dong Yan, Omar Hamed, Taruna Joshi,1 Mahmoud M. Mostafa, Kyla C. Jamieson, Radhika Joshi, Robert Newton, and Mark A. Giembycz Departments of Physiology and Pharmacology (D.Y., O.H., T.J., K.C.J., R.J., M.A.G.) and Cell Biology and Anatomy (M.M.M., R.N.), Snyder Institute for Chronic Diseases, Cumming School of Medicine, University of Calgary, Calgary, Alberta, Canada Received March 22, 2018; accepted April 11, 2018 Downloaded from ABSTRACT The contribution of gene expression changes to the adverse and activity, and positive regulation of neutrophil chemotaxis. The therapeutic effects of b2-adrenoceptor agonists in asthma was general enriched GO term extracellular space was also associ- investigated using human airway epithelial cells as a therapeu- ated with indacaterol-induced genes, and many of those, in- tically relevant target. Operational model-fitting established that cluding CRISPLD2, DMBT1, GAS1, and SOCS3, have putative jpet.aspetjournals.org the long-acting b2-adrenoceptor agonists (LABA) indacaterol, anti-inflammatory, antibacterial, and/or antiviral activity. Numer- salmeterol, formoterol, and picumeterol were full agonists on ous indacaterol-regulated genes were also induced or repressed BEAS-2B cells transfected with a cAMP-response element in BEAS-2B cells and human primary bronchial epithelial cells by reporter but differed in efficacy (indacaterol $ formoterol . -

Early Growth Response 1 Regulates Hematopoietic Support and Proliferation in Human Primary Bone Marrow Stromal Cells
Hematopoiesis SUPPLEMENTARY APPENDIX Early growth response 1 regulates hematopoietic support and proliferation in human primary bone marrow stromal cells Hongzhe Li, 1,2 Hooi-Ching Lim, 1,2 Dimitra Zacharaki, 1,2 Xiaojie Xian, 2,3 Keane J.G. Kenswil, 4 Sandro Bräunig, 1,2 Marc H.G.P. Raaijmakers, 4 Niels-Bjarne Woods, 2,3 Jenny Hansson, 1,2 and Stefan Scheding 1,2,5 1Division of Molecular Hematology, Department of Laboratory Medicine, Lund University, Lund, Sweden; 2Lund Stem Cell Center, Depart - ment of Laboratory Medicine, Lund University, Lund, Sweden; 3Division of Molecular Medicine and Gene Therapy, Department of Labora - tory Medicine, Lund University, Lund, Sweden; 4Department of Hematology, Erasmus MC Cancer Institute, Rotterdam, the Netherlands and 5Department of Hematology, Skåne University Hospital Lund, Skåne, Sweden ©2020 Ferrata Storti Foundation. This is an open-access paper. doi:10.3324/haematol. 2019.216648 Received: January 14, 2019. Accepted: July 19, 2019. Pre-published: August 1, 2019. Correspondence: STEFAN SCHEDING - [email protected] Li et al.: Supplemental data 1. Supplemental Materials and Methods BM-MNC isolation Bone marrow mononuclear cells (BM-MNC) from BM aspiration samples were isolated by density gradient centrifugation (LSM 1077 Lymphocyte, PAA, Pasching, Austria) either with or without prior incubation with RosetteSep Human Mesenchymal Stem Cell Enrichment Cocktail (STEMCELL Technologies, Vancouver, Canada) for lineage depletion (CD3, CD14, CD19, CD38, CD66b, glycophorin A). BM-MNCs from fetal long bones and adult hip bones were isolated as reported previously 1 by gently crushing bones (femora, tibiae, fibulae, humeri, radii and ulna) in PBS+0.5% FCS subsequent passing of the cell suspension through a 40-µm filter. -
![LC3B (MAP1LC3B) (N-Term) (Incl. Pos. Control) Mouse Monoclonal Antibody [Clone ID: 2G6] Product Data](https://docslib.b-cdn.net/cover/7264/lc3b-map1lc3b-n-term-incl-pos-control-mouse-monoclonal-antibody-clone-id-2g6-product-data-997264.webp)
LC3B (MAP1LC3B) (N-Term) (Incl. Pos. Control) Mouse Monoclonal Antibody [Clone ID: 2G6] Product Data
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for AM20213PU-N LC3B (MAP1LC3B) (N-term) (incl. pos. control) Mouse Monoclonal Antibody [Clone ID: 2G6] Product data: Product Type: Primary Antibodies Clone Name: 2G6 Applications: IF, WB Recommended Dilution: Immunoblotting: 0.5 µg/ml for HRPO/ECL detection Recommended blocking buffer: Casein/Tween 20 based blocking and blot incubation buffer. We strongly recommend to use PVDF membranes for immunoblot analysis. Immunocytochemistry: Use at 1-10 µg/ml (Paraformaldehyd/Methanol fixation). Included Positive Control: Cell lysate from untreated Neuro 2A (See Protocol below). Reactivity: Hamster, Human, Monkey, Mouse, Rat Host: Mouse Isotype: IgG1 Clonality: Monoclonal Immunogen: Synthetic peptide hemocyanin conjugated derived from the N-terminus of LC3-B Specificity: This antibody specifically recognizes both forms of endogenous LC3, the cytoplasmic LC3-I (18 kDa) as well as the lipidated form generated during autophagosome and autophagolysosome formation: LC3-II (16 kDa). Formulation: PBS containing 0.09% Sodium Azide, PEG and Sucrose/50% Glycerol State: Purified State: Liquid purified IgG fraction Concentration: lot specific Purification: Subsequent Ultrafiltration and Size Exclusion Chromatography Conjugation: Unconjugated Storage: Store the antibody (in aliquots) at -20°C. Avoid repeated freezing and thawing. Stability: Shelf life: one year from despatch. Gene Name: Homo sapiens microtubule associated protein 1 light chain 3 beta (MAP1LC3B) Database Link: Entrez Gene 64862 RatEntrez Gene 67443 MouseEntrez Gene 81631 Human Q9GZQ8 This product is to be used for laboratory only. Not for diagnostic or therapeutic use. -

Biological Models of Colorectal Cancer Metastasis and Tumor Suppression
BIOLOGICAL MODELS OF COLORECTAL CANCER METASTASIS AND TUMOR SUPPRESSION PROVIDE MECHANISTIC INSIGHTS TO GUIDE PERSONALIZED CARE OF THE COLORECTAL CANCER PATIENT By Jesse Joshua Smith Dissertation Submitted to the Faculty of the Graduate School of Vanderbilt University In partial fulfillment of the requirements For the degree of DOCTOR OF PHILOSOPHY In Cell and Developmental Biology May, 2010 Nashville, Tennessee Approved: Professor R. Daniel Beauchamp Professor Robert J. Coffey Professor Mark deCaestecker Professor Ethan Lee Professor Steven K. Hanks Copyright 2010 by Jesse Joshua Smith All Rights Reserved To my grandparents, Gladys and A.L. Lyth and Juanda Ruth and J.E. Smith, fully supportive and never in doubt. To my amazing and enduring parents, Rebecca Lyth and Jesse E. Smith, Jr., always there for me. .my sure foundation. To Jeannine, Bill and Reagan for encouragement, patience, love, trust and a solid backing. To Granny George and Shawn for loving support and care. And To my beautiful wife, Kelly, My heart, soul and great love, Infinitely supportive, patient and graceful. ii ACKNOWLEDGEMENTS This work would not have been possible without the financial support of the Vanderbilt Medical Scientist Training Program through the Clinical and Translational Science Award (Clinical Investigator Track), the Society of University Surgeons-Ethicon Scholarship Fund and the Surgical Oncology T32 grant and the Vanderbilt Medical Center Section of Surgical Sciences and the Department of Surgical Oncology. I am especially indebted to Drs. R. Daniel Beauchamp, Chairman of the Section of Surgical Sciences, Dr. James R. Goldenring, Vice Chairman of Research of the Department of Surgery, Dr. Naji N. -
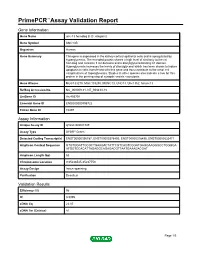
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name unc-13 homolog B (C. elegans) Gene Symbol UNC13B Organism Human Gene Summary This gene is expressed in the kidney cortical epithelial cells and is upregulated by hyperglycemia. The encoded protein shares a high level of similarity to the rat homolog and contains 3 C2 domains and a diacylglycerol-binding C1 domain. Hyperglycemia increases the levels of diacylglycerol which has been shown to induce apoptosis in cells transfected with this gene and thus contribute to the renal cell complications of hyperglycemia. Studies in other species also indicate a role for this protein in the priming step of synaptic vesicle exocytosis. Gene Aliases MGC133279, MGC133280, MUNC13, UNC13, Unc13h2, hmunc13 RefSeq Accession No. NC_000009.11, NT_008413.18 UniGene ID Hs.493791 Ensembl Gene ID ENSG00000198722 Entrez Gene ID 10497 Assay Information Unique Assay ID qHsaCID0007349 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000396787, ENST00000378495, ENST00000378496, ENST00000535471 Amplicon Context Sequence GTGTGGATTGCGCTGAAGACTATTCGTCAGTCGGATGAGGAAGGGCCTGGGGA ATGGTCCACATTAGAGGCAGAGACGTTAATGAAAGACGAT Amplicon Length (bp) 63 Chromosome Location 9:35236545-35237753 Assay Design Intron-spanning Purification Desalted Validation Results Efficiency (%) 96 R2 0.9995 cDNA Cq 22.07 cDNA Tm (Celsius) 81 Page 1/5 PrimePCR™Assay Validation Report gDNA Cq Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 2/5 PrimePCR™Assay Validation Report