Evolution of the First Metabolic Cycles
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Lipid Analysis of CO2-Rich Subsurface Aquifers Suggests an Autotrophy-Based Deep Biosphere with Lysolipids Enriched in CPR Bacteria
The ISME Journal (2020) 14:1547–1560 https://doi.org/10.1038/s41396-020-0624-4 ARTICLE Lipid analysis of CO2-rich subsurface aquifers suggests an autotrophy-based deep biosphere with lysolipids enriched in CPR bacteria 1,2 3,4 1,3 3 3 Alexander J. Probst ● Felix J. Elling ● Cindy J. Castelle ● Qingzeng Zhu ● Marcus Elvert ● 5,6 6 1 7,9 7 Giovanni Birarda ● Hoi-Ying N. Holman ● Katherine R. Lane ● Bethany Ladd ● M. Cathryn Ryan ● 8 3 1 Tanja Woyke ● Kai-Uwe Hinrichs ● Jillian F. Banfield Received: 20 November 2018 / Revised: 5 February 2020 / Accepted: 25 February 2020 / Published online: 13 March 2020 © The Author(s) 2020. This article is published with open access Abstract Sediment-hosted CO2-rich aquifers deep below the Colorado Plateau (USA) contain a remarkable diversity of uncultivated microorganisms, including Candidate Phyla Radiation (CPR) bacteria that are putative symbionts unable to synthesize membrane lipids. The origin of organic carbon in these ecosystems is unknown and the source of CPR membrane lipids remains elusive. We collected cells from deep groundwater brought to the surface by eruptions of Crystal Geyser, sequenced 1234567890();,: 1234567890();,: the community, and analyzed the whole community lipidome over time. Characteristic stable carbon isotopic compositions of microbial lipids suggest that bacterial and archaeal CO2 fixation ongoing in the deep subsurface provides organic carbon for the complex communities that reside there. Coupled lipidomic-metagenomic analysis indicates that CPR bacteria lack complete lipid biosynthesis pathways but still possess regular lipid membranes. These lipids may therefore originate from other community members, which also adapt to high in situ pressure by increasing fatty acid unsaturation. -

Process for Purification of Ethylene Compound Having Fluorine-Containing Organic Group
Office europeen des brevets (fi) Publication number : 0 506 374 A1 @ EUROPEAN PATENT APPLICATION @ Application number: 92302586.0 @ Int. CI.5: C07C 17/38, C07C 21/18 (g) Date of filing : 25.03.92 (30) Priority : 26.03.91 JP 86090/91 (72) Inventor : Kishita, Hirofumi 3-19-1, Isobe, Annaka-shi Gunma-ken (JP) (43) Date of publication of application : Inventor : Sato, Shinichi 30.09.92 Bulletin 92/40 3-5-5, Isobe, Annaka-shi Gunma-ken (JP) Inventor : Fujii, Hideki (84) Designated Contracting States : 3-12-37, Isobe, Annaka-shi DE FR GB Gunma-ken (JP) Inventor : Matsuda, Takashi 791 ~4 YdndSG Anri3k3~shi @ Applicant : SHIN-ETSU CHEMICAL CO., LTD. Gunma-ken (JP) 6-1, Ohtemachi 2-chome Chiyoda-ku Tokyo (JP) @) Representative : Votier, Sidney David et al CARPMAELS & RANSFORD 43, Bloomsbury Square London WC1A 2RA (GB) (54) Process for purification of ethylene compound having fluorine-containing organic group. (57) A process for purifying an ethylene compound having a fluorine-containing organic group (fluorine- containing ethylene compound) by mixing the fluorine-containing ethylene compound with an alkali metal or alkaline earth metal reducing agent, and subjecting the resulting mixture to irradiation with ultraviolet radiation, followed by washing with water. The purification process ensures effective removal of iodides which are sources of molecular iodine, from the fluorine-containing ethylene compound. < h- CO CO o 10 o Q_ LU Jouve, 18, rue Saint-Denis, 75001 PARIS EP 0 506 374 A1 BACKGROUND OF THE INVENTION 1. Field of the Invention 5 The present invention relates to a process for purifying an ethylene compound having a fluorine-containing organic group, and more particularly to a purification process by which iodine contained as impurity in an ethylene compound having a fluorine-containing organic group can be removed effectively. -
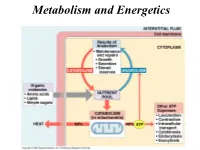
Metabolism and Energetics Oxidation of Carbon Atoms of Glucose Is the Major Source of Energy in Aerobic Metabolism
Metabolism and Energetics Oxidation of carbon atoms of glucose is the major source of energy in aerobic metabolism C6H1206 + 6O2 yields 6 CO2 + H20 + energy Energy released ΔG = - 686 kcal/mol Glucose oxidation requires over 25 discrete steps, with production of 36 ATP. Energy Transformations The mitochondrial synthesis of ATP is not stochiometric. Electron–motive force Proton-motive force Phosphoryl-transfer potential in the form of ATP. Substrate level phosphorylation The formation of ATP by substrate-level phosphorylation ADP ATP P CH O P CH2 O P 2 is used to represent HC OH HC OH a phosphate ester: phosphoglycerate CH OH OH CH2 O P kinase 2 P O bisphosphoglycerate 3-phosphoglycerate OH ADP ATP CH2 CH2 CH3 C O P C OH C O pyruvate kinase non-enzymic COOH COOH COOH phosphoenolpyruvate enolpyruvate pyruvate Why ATP? The reaction of ATP hydrolysis is very favorable ΔGo = - 30.5 kJ/mol = - 7.3 kCal/mol 1. Charge separation of closely packed phosphate groups provides electrostatic relief. Mg2+ 2. Inorganic Pi, the product of the reaction, is immediately resonance-stabilized (electron density spreads equally to all oxygens). 3. ADP immediately ionizes giving H+ into a low [H+] environment (pH~7). 4. Both Pi and ADP are more favorably solvated by water than one ATP molecule. 5. ATP is water soluble. The total body content of ATP and ADP is under 350 mmol – about 10 g, BUT … the amount of ATP synthesized and used each day is about 150 mol – about 110 kg. ATP Production - stage 1 - Glycolysis Glycolysis When rapid production of ATP is needed. -

Carlson Udel 0060D 13047.Pdf
SYNTHETIC CARBON FIXATION FOR IMPROVED MICROBIAL FERMENTATION YIELDS by Ellinor Dorothee Carlson A dissertation submitted to the Faculty of the University of Delaware in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Chemical Engineering Summer 2017 © 2017 Ellinor Dorothee Carlson All Rights Reserved SYNTHETIC CARBON FIXATION FOR IMPROVED MICROBIAL FERMENTATION YIELDS by Ellinor Dorothee Carlson Approved: __________________________________________________________ Abraham M. Lenhoff, Ph.D. Chair of the Department of Chemical & Biomolecular Engineering Approved: __________________________________________________________ Babatunde A. Ogunnaike, Ph.D. Dean of the College of Engineering Approved: __________________________________________________________ Ann L. Ardis, Ph.D. Senior Vice Provost for Graduate and Professional Education I certify that I have read this dissertation and that in my opinion it meets the academic and professional standard required by the University as a dissertation for the degree of Doctor of Philosophy. Signed: __________________________________________________________ Eleftherios T. Papoutsakis, Ph.D. Professor in charge of dissertation I certify that I have read this dissertation and that in my opinion it meets the academic and professional standard required by the University as a dissertation for the degree of Doctor of Philosophy. Signed: __________________________________________________________ Maciek R. Antoniewicz, Ph.D. Member of dissertation committee I certify that I have read this dissertation and that in my opinion it meets the academic and professional standard required by the University as a dissertation for the degree of Doctor of Philosophy. Signed: __________________________________________________________ Wilfred Chen, Ph.D. Member of dissertation committee I certify that I have read this dissertation and that in my opinion it meets the academic and professional standard required by the University as a dissertation for the degree of Doctor of Philosophy. -

Photosynthesis and Cellular Respiration
Unit 4 - Photosynthesis and Cellular Respiration Topic Products and Reactants Best and Worst Colors for Photos nthesis " Light Dependent vs. Light Independent Reaction Organelles Responsible ADP VS. ATP Products and Reactants Photosynthesis Cellular Respiration Aerobic: C6H120 6 + O2 ~C02 + H20 + 36 ATP Anaerobic: Human: C6H1206~ CO 2 + lactic acid + 4ATP • Yeast: C6H1206~ CO 2 + ethyl alcohol + 4ATP Best and Worst • Colors for . Photosynthesis i Light Dependent vs. Light Independent Reaction Organelles Responsible ADP vs. AlP 8986 - 1 - Page 1 Nanre: _______________________________________ In animal cells, the energy to convert ADP to AlP comes directly from A) organic molecules C) sunlight B) inorganic molecules D) hormones 2) Which statement correctly describes part ofthe photosynthetic process in plants? A) Water is spili in the light reactions. B) Alcohol is produced by the light reactions. C) Oxygen is used in the dark reactions. D) Carbon dioxide is released in the dark reactions. 3) Which statement best describes one ofthe events taking p1ace in the ,chemical reaction represented below? ATPase H 0 + ATP --------~) ADP + P + energy 2 A) Photosynthesis is taking place, resulting in the storage ofenergy. B) Energy is being stored as a result ofaerobic respiratioIt C) Energy is being released fur metabolic activities. D) Fermentation is taking place, resulting in the synthesis ofAlP. 4) Which process results in muscle fatigue and cramping in humans? A) aerobic respiration C) lactic acid fermentation B) photosynthesis D) alcoholic fermentation -

Reductions and Reducing Agents
REDUCTIONS AND REDUCING AGENTS 1 Reductions and Reducing Agents • Basic definition of reduction: Addition of hydrogen or removal of oxygen • Addition of electrons 9:45 AM 2 Reducible Functional Groups 9:45 AM 3 Categories of Common Reducing Agents 9:45 AM 4 Relative Reactivity of Nucleophiles at the Reducible Functional Groups In the absence of any secondary interactions, the carbonyl compounds exhibit the following order of reactivity at the carbonyl This order may however be reversed in the presence of unique secondary interactions inherent in the molecule; interactions that may 9:45 AM be activated by some property of the reacting partner 5 Common Reducing Agents (Borohydrides) Reduction of Amides to Amines 9:45 AM 6 Common Reducing Agents (Borohydrides) Reduction of Carboxylic Acids to Primary Alcohols O 3 R CO2H + BH3 R O B + 3 H 3 2 Acyloxyborane 9:45 AM 7 Common Reducing Agents (Sodium Borohydride) The reductions with NaBH4 are commonly carried out in EtOH (Serving as a protic solvent) Note that nucleophilic attack occurs from the least hindered face of the 8 carbonyl Common Reducing Agents (Lithium Borohydride) The reductions with LiBH4 are commonly carried out in THF or ether Note that nucleophilic attack occurs from the least hindered face of the 9:45 AM 9 carbonyl. Common Reducing Agents (Borohydrides) The Influence of Metal Cations on Reactivity As a result of the differences in reactivity between sodium borohydride and lithium borohydride, chemoselectivity of reduction can be achieved by a judicious choice of reducing agent. 9:45 AM 10 Common Reducing Agents (Sodium Cyanoborohydride) 9:45 AM 11 Common Reducing Agents (Reductive Amination with Sodium Cyanoborohydride) 9:45 AM 12 Lithium Aluminium Hydride Lithium aluminiumhydride reacts the same way as lithium borohydride. -

Chapter 20 Electrochemistry
Chapter 20 Electrochemistry Learning goals and key skills: Identify oxidation, reduction, oxidizing agent, and reducing agent in a chemical equation Complete and balance redox equations using the method of half-reactions. Sketch a voltaic cell and identify its cathode, anode, and the directions in which electrons and ions move. o Calculate standard emfs (cell potentials), E cell, from standard reduction potentials. Use reduction potentials to predict whether a redox reaction is spontaneous. o o Relate E cell to DG and equilibrium constants. Calculate emf under nonstandard conditions. Identify the components of common batteries. Describe the construction of a lithium-ion battery and explain how it works. Describe the construction of a fuel cell and explain how it generates electrical energy. Explain how corrosion occurs and how it is prevented by cathodic protection. Describe the reactions in electrolytic cells. Relate the amounts of products and reactants in redox reactions to electrical charge. Electrochemistry Electrochemistry is the study of the relationships between electricity and chemical reactions. • It includes the study of both spontaneous and nonspontaneous processes. 1 Redox reactions: assigning oxidation numbers Oxidation numbers help keep track of what species loses electrons and what species gains them. • An element is oxidized when the oxidation number increases • An element is reduced when the oxidation number decreases • an oxidizing agent causes another element to be oxidized • a reducing agent causes another element to be reduced. Assigning oxidation numbers (sect. 4.4) 1. Elemental form, each atom has ox. # = 0. Zn O2 O3 I2 S8 P4 2. Simple ions, = charge on ion. -1 for Cl-, +2 for Mg2+ 3. -

Ch. 21.1 Redox Reactions and Electrochemical Cells
Pre-Health Post-Baccalaureate Program Study Guide and Practice Problems Course: CHM2046 Textbook Chapter: 21.1 (Silberberg 6e) Topics Covered: Redox Reactions and Electrochemical Cells Created by Isaac Loy 1. Review Understanding this chapter’s material will depend on an in- depth understanding of redox reactions, which were first covered last semester in ch. 4. We will review redox reactions in this study guide, but it would be wise to review ch. 4 if you are having difficulty with this material. Redox reactions will also be incredibly important moving forward into organic chemistry and biochemistry. 2. Oxidation-Reduction Reactions The mnemonic that you will come back to time and time again for this topic is: “LEO the lion says GER” Where “LEO” stands for Loss of Electrons = Oxidation And “GER” stands for Gain of Electrons = Reduction The oxidizing agent (the substance that is being reduced) pulls electrons from the substance that is being oxidized. The reducing agent (the substance that is being oxidized) gives electrons to the substance that is being reduced. Oxidation and reduction are simultaneous processes. In order for a redox reaction to take place, one substance must be oxidized and the other must be reduced. When working with oxidation numbers to solve problems, the substance being oxidized (LEO -> loss of electrons) becomes more positive. Likewise, the substance being reduced (GER -> gaining electrons) becomes more negative. 3. Using Half-Reactions to Solve Redox Problems Follow the steps below to create half-reactions. It is crucial that you follow the steps in order! A. Split up the overall reaction into two half-reactions, where the species of one reaction is being oxidized, and the species of the other reaction is being reduced. -

Gen Chem II Jasperse Ch. 19 Electrochemistry 1
Gen Chem II Jasperse Ch. 19 Electrochemistry 1 Chapter 19 Electrochemistry Math Summary Relating Standard Cell Potential to Standard Half Cell Potentials Eºcell=Eºoxidation + Eºreduction (standard conditions assume 1.0 M concentrations) Relating Half Cell Potentials when Written in Opposite Directions Eºox = -Eºred for half reactions written in opposite directions Relating Standard Cell Potentials to ∆G ∆Gº = -nFE˚cell (to give answer in kJ, use F = 96.485) F = 96,500 C/mol n=number of electrons transferred Relating Actual Cell Potential to Standard Cell Potential when Concentrations aren't 1.0-M Ecell = Eºcell -[0.0592/n] log Q (Q = ratio of actual concentrations) Relating Standard Cell Potential to Equilibrium Constant log K = nEº/0.0592 Relating Actual Cell Potential to Actual Concentrations in Concentration Cells Ecell = -[0.0592/n] log Q for concentration cells, where anode and cathode differ only in concentration, but otherwise have same ions Relating # of Moles of Electrons Transferred as a Function of Time and Current in Electrolysis 1 mol e- = 96,500 C moles of electrons = [current (A)•time (sec)]/96,500 for electrolysis, moles, current, and time are related. rearranged: time (sec)=(moles of electrons)(96500)/current (in A) Note: 3600 sec/hour so time (hours)=(moles of electrons)(26.8)/current (in A) Electrochemistry-Related Units C = Coulomb = quantity of electrical charge = 6.24 • 1018 electrons • 1 mole of electrons = 96,500 C A = amp = rate of charge flow per time = C/sec V = volt = electrical power/force/strength = J/C 96,500C 96.5 kJ F = Faraday = = mole e− mole e− •V € € Gen Chem II Jasperse Ch. -

The Formation of a Gluconeogenesis System and Reductive Pentose Phosphate Pathway of CO2 Fixation in Ancient Hydrothermal Systems Sergey A
: O tics pe ge n r A e c n c e Marakushev and Belonogova, Bioenergetics 2016, e o s i s B 5:2 Bioenergetics: Open Access DOI: 10.4172/2167-7662.1000141 ISSN: 2167-7662 Research Article Open Access The Emergence of Bioenergetics: The Formation of a Gluconeogenesis System and Reductive Pentose Phosphate Pathway of CO2 Fixation in Ancient Hydrothermal Systems Sergey A. Marakushev* and Ol’ga V. Belonogova Institute of Problems of Chemical Physics, Russian Academy of Sciences, Chernogolovka, Moscow Region, Russia *Corresponding author: Marakushev SA, Institute of Problems of Chemical Physics, Russian Academy of Sciences, Chernogolovka, Russia, Tel: 496-522-7772; E-mail: [email protected] Rec date: Apr 18, 2016; Acc date: Nov 08, 2016; Pub date: Nov 11, 2016 Copyright: © 2016 Marakushev SA et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. Abstract The origin of phosphorus metabolism is one of the central problems in the context of the emergence of life on Earth. It has been shown that the C–H–O system can be transformed into a four- component C–H–O–P system with the formation of a gluconeogenesis path in a possible Archean hydrothermal condition under the influence of a phosphorus chemical potential. This system became the energy supply basis for protometabolism, and facilitated the formation of a new CO2 fixation cycle (the reductive pentose phosphate pathway). The modular design of central metabolism in the C–H–O–P system is derived from the parageneses (associations) of certain substances, and the emerging modules in turn associate with each other in certain physical and chemical hydrothermal conditions. -

Direction of Krebs Cycle Which Way Does the Citric Acid Cycle
Direction of Krebs cycle Which way does the citric acid cycle turn during hypoxia? The critical role of alpha-ketoglutarate dehydrogenase complex Christos Chinopoulos Department of Medical Biochemistry, Semmelweis University, Budapest, 1094, Hungary Running title: Direction of Krebs cycle Address correspondence to: Dr. Christos Chinopoulos, Department of Medical Biochemistry, Semmelweis University, Budapest, Hungary. Tel: +361 4591500 ext. 60024, Fax: +361 2670031. E-mail: [email protected] Grant information: Work cited from the author's laboratory was supported by the Országos Tudományos Kutatási Alapprogram (OTKA) grants NNF 78905, NNF2 85658, K 100918 and the MTA-SE Lendület Neurobiochemistry Research Division grant 95003. 1 Direction of Krebs cycle Abstract The citric acid cycle forms a major metabolic hub and as such it is involved in many disease states implicating energetic imbalance. In spite of the fact that it is being branded as a 'cycle', during hypoxia, when the electron transport chain does not oxidize reducing equivalents, segments of this metabolic pathway remain operational but exhibit opposing directionalities. This serves the purpose of harnessing high energy phosphates through matrix substrate-level phosphorylation in the absence of oxidative phosphorylation. In this mini-review these segments are appraised, pointing to the critical importance of the alpha-ketoglutarate dehydrogenase complex dictating their directionalities. Keywords: tricarboxylic acid cycle; Krebs cycle; energy metabolism; mitochondria, cancer 2 Direction of Krebs cycle Background The citric acid cycle (Krebs, 1940a) consists of sequential, reversible and irreversible biochemical reactions, shown in figure 1. There are many 'entry' points to this pathway but as the name 'cycle' implies, there is no end: every product of a reaction is a substrate for the next one eventually leading to the regeneration of each one of them. -

Coenzyme a Carboxylase-Α Gene Transcription by the Liver X Receptor Agonist T0-901317 Saswata Talukdar
Faculty Scholarship 2006 The mechanism mediating the activation of acetyl- coenzyme A carboxylase-α gene transcription by the liver X receptor agonist T0-901317 Saswata Talukdar F. Bradley Hillgartner Follow this and additional works at: https://researchrepository.wvu.edu/faculty_publications Digital Commons Citation Talukdar, Saswata and Hillgartner, F. Bradley, "The mechanism mediating the activation of acetyl-coenzyme A carboxylase-α gene transcription by the liver X receptor agonist T0-901317" (2006). Faculty Scholarship. 377. https://researchrepository.wvu.edu/faculty_publications/377 This Article is brought to you for free and open access by The Research Repository @ WVU. It has been accepted for inclusion in Faculty Scholarship by an authorized administrator of The Research Repository @ WVU. For more information, please contact [email protected]. The mechanism mediating the activation of acetyl- coenzyme A carboxylase-a gene transcription by the liver X receptor agonist T0-901317 Saswata Talukdar and F. Bradley Hillgartner1 Department of Biochemistry and Molecular Pharmacology, School of Medicine, West Virginia University, Morgantown, WV 26506 Abstract In birds and mammals, agonists of the liver X re- elongases involved in the chain elongation of fatty acids to ceptor (LXR) increase the expression of enzymes that make very-long-chain fatty acids (3). The essential role of ACCa up the fatty acid synthesis pathway. Here, we investigate the in lipid biosynthesis has been confirmed by studies dem- mechanism by which the synthetic LXR agonist, T0-901317, onstrating that knockout of the ACCa gene disrupts em- increases the transcription of the acetyl-coenzyme A carbox- ylase-a (ACCa) gene in chick embryo hepatocyte cultures.