Exome Analysis Identifies Brody Myopathy in a Family Diagnosed
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Ebf Factors and Myod Cooperate to Regulate Muscle Relaxation Via Atp2a1
ARTICLE Received 12 Feb 2014 | Accepted 2 Apr 2014 | Published 2 May 2014 DOI: 10.1038/ncomms4793 Ebf factors and MyoD cooperate to regulate muscle relaxation via Atp2a1 Saihong Jin1, Jeehee Kim1, Torsten Willert1, Tanja Klein-Rodewald2, Mario Garcia-Dominguez3,4, Matias Mosqueira5, Rainer Fink5, Irene Esposito2,6, Lorenz C. Hofbauer7, Patrick Charnay3 & Matthias Kieslinger1 Myogenic regulatory factors such as MyoD and Myf5 lie at the core of vertebrate muscle differentiation. However, E-boxes, the cognate binding sites for these transcription factors, are not restricted to the promoters/enhancers of muscle cell-specific genes. Thus, the specificity in myogenic transcription is poorly defined. Here we describe the transcription factor Ebf3 as a new determinant of muscle cell-specific transcription. In the absence of Ebf3 the lung does not unfold at birth, resulting in respiratory failure and perinatal death. This is due to a hypercontractile diaphragm with impaired Ca2 þ efflux-related muscle functions. Expression of the Ca2 þ pump Serca1 (Atp2a1) is downregulated in the absence of Ebf3, and its transgenic expression rescues this phenotype. Ebf3 binds directly to the promoter of Atp2a1 and synergises with MyoD in the induction of Atp2a1. In skeletal muscle, the homologous family member Ebf1 is strongly expressed and together with MyoD induces Atp2a1. Thus, Ebf3 is a new regulator of terminal muscle differentiation in the diaphragm, and Ebf factors cooperate with MyoD in the induction of muscle-specific genes. 1 Institute of Clinical Molecular Biology and Tumor Genetics, Helmholtz Zentrum Mu¨nchen, National Research Center for Environmental Health, Marchioninistrasse 25, 81377 Munich, Germany. 2 Institute of Pathology, Helmholtz Zentrum Mu¨nchen, National Research Center for Environmental Health, Ingolsta¨dter Landstr. -

ATP2A1 Gene Atpase Sarcoplasmic/Endoplasmic Reticulum Ca2+ Transporting 1
ATP2A1 gene ATPase sarcoplasmic/endoplasmic reticulum Ca2+ transporting 1 Normal Function The ATP2A1 gene provides instructions for making an enzyme called sarco(endo) plasmic reticulum calcium-ATPase 1 (SERCA1). This enzyme belongs to a family of ATPase enzymes that help control the level of positively charged calcium atoms ( calcium ions) inside cells. The SERCA1 enzyme is found in skeletal muscle cells. ( Skeletal muscles are the muscles used for movement.) Within muscle cells, the SERCA1 enzyme is located in the membrane of a structure called the sarcoplasmic reticulum. This structure plays a major role in muscle contraction and relaxation by storing and releasing calcium ions. When calcium ions are transported out of the sarcoplasmic reticulum, muscles contract; when calcium ions are transported into the sarcoplasmic reticulum, muscles relax. The SERCA1 enzyme transports calcium ions from the cell into the sarcoplasmic reticulum, triggering muscle relaxation. Health Conditions Related to Genetic Changes Brody myopathy At least 10 mutations in the ATP2A1 gene have been found to cause Brody myopathy, a muscle disorder characterized by muscle cramping after exercise. Most ATP2A1 gene mutations lead to a premature stop signal in the instructions for making the SERCA1 enzyme, resulting in a nonfunctional enzyme. Other mutations lead to the production of a SERCA1 enzyme with decreased function. As a result, calcium ions are slow to enter the sarcoplasmic reticulum and muscle relaxation is delayed. After exercise or other strenuous activity, during which the muscles rapidly contract and relax, people with Brody myopathy develop muscle cramps because their muscles cannot fully relax. Scientists believe that other proteins or other pathways may function in the absence of a fully functional SERCA1 enzyme to transport calcium ions into the sarcoplasmic reticulum and help with muscle relaxation. -

WO 2015/048577 A2 April 2015 (02.04.2015) W P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2015/048577 A2 April 2015 (02.04.2015) W P O P C T (51) International Patent Classification: (81) Designated States (unless otherwise indicated, for every A61K 48/00 (2006.01) kind of national protection available): AE, AG, AL, AM, AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, (21) International Application Number: BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, PCT/US20 14/057905 DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, (22) International Filing Date: HN, HR, HU, ID, IL, IN, IR, IS, JP, KE, KG, KN, KP, KR, 26 September 2014 (26.09.2014) KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, (25) Filing Language: English PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, SC, (26) Publication Language: English SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (30) Priority Data: 61/883,925 27 September 2013 (27.09.2013) US (84) Designated States (unless otherwise indicated, for every 61/898,043 31 October 2013 (3 1. 10.2013) US kind of regional protection available): ARIPO (BW, GH, GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, ST, SZ, (71) Applicant: EDITAS MEDICINE, INC. -

Identification of a Missense Mutation in the Bovine ATP2A1 Gene
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Genomics 92 (2008) 474–477 Contents lists available at ScienceDirect Genomics journal homepage: www.elsevier.com/locate/ygeno Identification of a missense mutation in the bovine ATP2A1 gene in congenital pseudomyotonia of Chianina cattle: An animal model of human Brody disease Cord Drögemüller a,⁎, Michaela Drögemüller a, Tosso Leeb a, Francesco Mascarello b, Stefania Testoni c, Marco Rossi d, Arcangelo Gentile d, Ernesto Damiani e, Roberta Sacchetto b a Institute of Genetics, Vetsuisse Faculty, University of Berne, Bremgartenstrasse 109a, CH-3001 Berne, Switzerland b Department of Experimental Veterinary Sciences, University of Padua, Italy c Department of Veterinary Clinical Sciences, University of Padua, Italy d Veterinary Clinical Department, University of Bologna, Italy e Department of Experimental Biomedical Sciences, University of Padua, Italy article info abstract Article history: Congenital pseudomyotonia in Chianina cattle is a muscle function disorder very similar to that of Brody Received 6 July 2008 disease in humans. Mutations in the human ATP2A1 gene, encoding SERCA1, cause Brody myopathy. The Accepted 31 July 2008 analysis of the collected Chianina pedigree data suggested monogenic autosomal recessive inheritance and Available online 25 September 2008 revealed that all 17 affected individuals traced back to a single founder. A deficiency of SERCA1 function in skeletal muscle of pseudomyotonia affected Chianina cattle was observed as SERCA1 activity in affected Keywords: Cattle animals was decreased by about 70%. Linkage analysis showed that the mutation was located in the ATP2A1 Chianina gene region on BTA25 and subsequent mutation analysis of the ATP2A1 exons revealed a perfectly associated ATP2A1 missense mutation in exon 6 (c.491GNA) leading to a p.Arg164His substitution. -

Download CGT Exome V2.0
CGT Exome version 2. -

ICNMD XIII 13Th International Congress on Neuromuscular
ICNMD XIII 13th International congress on Neuromuscular Diseases Nice, France July 5-10, 2014 Poster Sessions Abstract Books Journal of Neuromuscular Diseases 1 (2014) S83–S403 S83 DOI 10.3233/JND-149002 IOS Press Abstracts PF1 These unexpected results were confi rmed in vitro in cultured myoblasts. Accordingly MyoD and MyoG expressions were not altered by Srf loss. PS1-1 / #138 - that SC late differentiation and fi ber growth were Theme: 1.1 - Basic sciences in NMD: Muscle homeostasis / Muscle altered in vivo and in vitro. regeneration Further characterization of SC cell behaviour (self- renewal, motility, survival...) will be presented. In ad- Role of Serum response factor in dition, we performed transcriptomic studies and muscular satellite cells identifi ed the set of genes whose expressions are al- tered by Srf loss in proliferating and differentiating Voahangy Randrianarison-Huetz, Aikaterini myoblasts. Genes identifi ed in this screen will aid de- Papaefthymiou, Laura Collard, Ulduz Faradova, ciphering the underlying molecular mechanism. Athanassia Sotiropoulos Genetics and Development, Institut Cochin, Paris, France PS1-2 / #247 In the muscular system, thetranscription factor Srf Theme: 1.1 - Basic sciences in NMD: Muscle homeostasis / Muscle (Serum response factor) controls the expression of a regeneration wide range of genes including those involved in pro- liferation (immediate early genes) and myogenic dif- Sdf-1 promotes BMSCs participation in ferentiation (MyoD, a-actin). Indeed, inhibition of Srf regeneration of Pax7-/- mouse skeletal in cultured myogenic cell lines C2C12 was shown to impede myoblast’s proliferation and differentiation muscles into myotubes. However data are lacking regarding Kamil Kowalski, Maria Ciemerych, Edyta Brzóska the role of Srf in muscle stem cells behavior in vivo. -

Neuro)Muscular Panel 03-12-2018 Versie Centrum Voor Medische Genetica Gent (215 Genen
H9.1-OP2-B9: Genpanel Neuromusculaire Dystrofie, 03-12-2018 in voege op 3/12/2018 (Neuro)Muscular panel 03-12-2018 versie Centrum voor Medische Genetica Gent (215 genen) GitHub commit: e0a58ebf6cd19fa332a01b6b7109c050aee29f3e Associated phenotype, OMIM phenotype ID, phenotype Gene OMIM gene ID mapping key and inheritance pattern ABHD5 604780 Chanarin-Dorfman syndrome, 275630 (3), Autosomal recessive ACADVL 609575 VLCAD deficiency, 201475 (3), Autosomal recessive Myopathy, actin, congenital, with cores, 161800 (3), Autosomal recessive, Autosomal dominant; Myopathy, actin, congenital, with excess of thin myofilaments, 161800 (3), Autosomal recessive, Autosomal dominant; Myopathy, congenital, with fiber-type ACTA1 102610 disproportion 1, 255310 (3), Autosomal recessive, Autosomal dominant; ?Myopathy, scapulohumeroperoneal, 616852 (3), Autosomal dominant; Nemaline myopathy 3, autosomal dominant or recessive, 161800 (3), Autosomal recessive, Autosomal dominant Fibrodys plasia ossificans progressiva, 135100 (3), Autosomal ACVR1 102576 dominant Glycogen storage disease IIIa, 232400 (3), Autosomal recessive; AGL 610860 Glycogen storage disease IIIb, 232400 (3), Autosomal recessive Myasthenic syndrome, congenital, 8, with pre- and postsynaptic AGRN 103320 defects, 615120 (3), Autosomal recessive ALDOA 103850 Glycogen storage disease XII, 611881 (3), Autosomal recessive ?Congenital disorder of glycosylation, type Is, 300884 (3), X -linked ALG13 300776 dominant; Epileptic encephalopathy, early infantile, 36, 300884 (3), X-linked dominant ?Myasthenic -
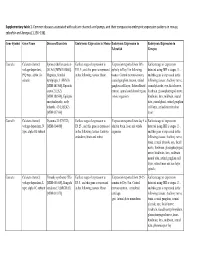
Supplementary Tables 1 Final.Xlsx
Supplementary table 1. Common diseases associated with calcium channels and pumps, and their comparative embryonic expression patterns in mouse, zebrafish and Xenopus [1,136–138]. Gene Symbol Gene Name Diseases/Disorders Embryonic Expression in Mouse Embryonic Expression in Embryonic Expression in Zebrafish Xenopus Cacna1a Calcium channel, Spinocerebellar ataxia 6 Earliest stage of expression is Expression reported from 50% Earliest stage of expression voltage-dependent, (SCA6) [MIM:183086], E11.5 , and this gene is expressed epiboly to Day 5 in following detected using ISH is stages 13 , P/Q type, alpha 1A Migraine, familial in the following tissues: Brain tissues: Central nervous system, and this gene is expressed in the subunit hemiplegic, 1 (FHM1) cranial ganglion, neuron, retinal following tissues: Auditory nerve, [MIM:141500], Episodic ganglion cell layer, Rohon-Beard cranial placode, eye, facial nerve, ataxia 2 (EA2) neuron, spinal cord dorsal region, forebrain, glossopharyngeal nerve, [MIM:108500], Epileptic whole organism hindbrain, lens, midbrain, neural encephalopathy, early tube, pineal gland, retinal ganglion infantile, 42 (EIEE42) cell layer, retinal inner nuclear [MIM:617106] layer Cacna1b Calcium channel, Dystonia 23 (DYT23) Earliest stage of expression is Expression reported from day 5 to Earliest stage of expression voltage-dependent, N [MIM:614860] E8.25 , and this gene is expressed adult in brain, liver and whole detected using ISH is stages 13 , type, alpha 1B subunit in the following tissues: Embryo organism and this -

Genomeposter2009.Pdf
Fold HumanSelected Genome Genes, Traits, and Landmarks Disorders www.ornl.gov/hgmis/posters/chromosome genomics.energy. -

Primary Active Ca2+ Transport Systems in Health and Disease
Downloaded from http://cshperspectives.cshlp.org/ on September 27, 2021 - Published by Cold Spring Harbor Laboratory Press Primary Active Ca2+ Transport Systems in Health and Disease Jialin Chen,1 Aljona Sitsel,1 Veronick Benoy,1 M. Rosario Sepúlveda,2,3 and Peter Vangheluwe1,3 1Laboratory of Cellular Transport Systems, Department of Cellular and Molecular Medicine, KU Leuven, 3000 Leuven, Belgium 2Department of Cell Biology, Faculty of Sciences, University of Granada, 18071 Granada, Spain Correspondence: [email protected] Calcium ions (Ca2+) are prominent cell signaling effectors that regulate a wide variety of cellular processes. Among the different players in Ca2+ homeostasis, primary active Ca2+ transporters are responsible for keeping low basal Ca2+ levels in the cytosol while establishing steep Ca2+ gradients across intracellular membranes or the plasma membrane. This review summarizes our current knowledge on the three types of primary active Ca2+-ATPases: the sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA) pumps, the secretory pathway Ca2+- ATPase (SPCA) isoforms, and the plasma membrane Ca2+-ATPase (PMCA) Ca2+-transporters. We first discuss the Ca2+ transport mechanism of SERCA1a, which serves as a reference to describe the Ca2+ transport of other Ca2+ pumps. We further highlight the common and unique features of each isoform and review their structure–function relationship, expression pattern, regulatory mechanisms, and specific physiological roles. Finally, we discuss the increasing genetic and in vivo evidence that links the dysfunction of specific Ca2+-ATPase isoforms to a broad range of human pathologies, and highlight emerging therapeutic strate- gies that target Ca2+ pumps. a2+ signaling is crucial for many physiolog- cus on the primary active Ca2+-transporters or Cical processes and is dysregulated in a mul- Ca2+-ATPases, which are responsible for keep- titude of pathological conditions. -

GENETIC TESTING REQUISITION Please Ship All NON-PRENATAL
GENETIC TESTING REQUISITION 1-844-363-4357· [email protected] Schillingallee 68 · 18057 Rostock Germany Attention Patient: Please visit your nearest LifeLabs or CML Healthcare Patient Service Centre for sample collection CONTRACT # LL: K012-01/ CML: CEN Report to Physician Billing # LifeLabs Demographic Label Ordering Physician Name Physician Signature: Ordering Physician Address: Tel: Fax: Address & Contact Info: Copy to (name & contact info): Name: Contact: Bill to Contract # K012-01 (patient does not pay at time of collection) Patient Gender: (M/F) Patient Name (Last, First): Patient DOB: (YYYY/MM/DD) Patient Address: Patient Health Card: Patient Telephone: Please ship all NON-PRENATAL samples to: LifeLabs · Attn CDS Department • 100 International Boulevard• Toronto ON• M9W6J6 TEST REQUESTED LL TR # / CML TC# □ Genetic Test - Blood Sample 2 x 4mL EDTA 4005 □ Genetic Test (Pediatric) - Blood Sample 1 x 2mL EDTA 4008 □ Genetic Test - Other Sample Type 4014 PRENATAL SAMPLES: Please ship directly to CENTOGENE. Date Blood Collected (YYYY/MM/DD): ___________ Time Blood Collected (HH:MM)) :________ Collector Name: - ___________________ GENETIC TESTING CONSENT I understand that a DNA specimen will be sent to LifeLabs for genetic testing. My physician has told me about the condition(s) being tested and its genetic basis. I am aware that correct information about the relationships between my family members is important. I agree that my specimen and personal health information may be sent to Centogene AG at their lab in Germany (address below). To ensure accurate testing, I agree that the results of any genetic testing that I have had previously completed by Centogene AG may be shared with LifeLabs. -

Nuove Politiche Per L'innovazione Nel Settore Delle Scienze Della Vita
Laura Magazzini Fabio Pammolli Massimo Riccaboni WP CERM 03-2009 NUOVE POLITICHE PER L'INNOVAZIONE NEL SETTORE DELLE SCIENZE DELLA VITA ISBN 978-88-3289-038-9 INDICE EXECUTIVE SUMMARY .................................................................................. 2 1. Risorse e innovazione: fallimenti di mercato e logiche di intervento pubblico........... 2 2. Da raro a generale: nuovi modelli di sostegno mission-oriented alla ricerca e sviluppo nelle scienze della vita............................................................................... 31 2.1. Incentivi pubblici per la ricerca sulle malattie rare: il panorama internazionale.....37 Stati Uniti...........................................................................................................................................................................................37 Giappone.............................................................................................................................................................................................44 Australia..............................................................................................................................................................................................46 Unione Europea.............................................................................................................................................................................46 2.2. Incentivi pubblici per la ricerca sulle malattie rare: il panorama europeo.....................58 Francia ..................................................................................................................................................................................................58