DGCR6 at the Proximal Part of the Digeorge Critical Region Is Involved in Conotruncal Heart Defects
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
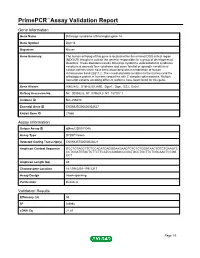
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name DiGeorge syndrome critical region gene 14 Gene Symbol Dgcr14 Organism Mouse Gene Summary The human ortholog of this gene is located within the minimal DGS critical region (MDGCR) thought to contain the gene(s) responsible for a group of developmental disorders. These disorders include DiGeorge syndrome velocardiofacial syndrome conotruncal anomaly face syndrome and some familial or sporadic conotruncal cardiac defects which have been associated with microdeletion of human chromosome band 22q11.2. The encoded protein localizes to the nucleus and the orthologous protein in humans co-purifies with C complex spliceosomes. Multiple transcript variants encoding different isoforms have been found for this gene. Gene Aliases AI462402, D16H22S1269E, Dgcr1, Dgsi, ES2, Es2el RefSeq Accession No. NC_000082.6, NT_039624.8, NT_187007.1 UniGene ID Mm.256480 Ensembl Gene ID ENSMUSG00000003527 Entrez Gene ID 27886 Assay Information Unique Assay ID qMmuCID0011048 Assay Type SYBR® Green Detected Coding Transcript(s) ENSMUST00000003621 Amplicon Context Sequence GCCTGTAGCTTCTCCACATCAGGGAAGAAGTCTCTCTGGATAACTGTCTGAAGTC CCTCGATGTACTCTTCTTCATCCAGGACCCGCTGCCTGCTTCTCGCAACTCCGG CCT Amplicon Length (bp) 82 Chromosome Location 16:17910201-17911217 Assay Design Intron-spanning Purification Desalted Validation Results Efficiency (%) 95 R2 0.9994 cDNA Cq 21.87 Page 1/5 PrimePCR™Assay Validation Report cDNA Tm (Celsius) 79.5 gDNA Cq 23.94 Specificity (%) 100 Information to assist with data interpretation is provided -

CRNKL1 Is a Highly Selective Regulator of Intron-Retaining HIV-1 and Cellular Mrnas
bioRxiv preprint doi: https://doi.org/10.1101/2020.02.04.934927; this version posted February 11, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 CRNKL1 is a highly selective regulator of intron-retaining HIV-1 and cellular mRNAs 2 3 4 Han Xiao1, Emanuel Wyler2#, Miha Milek2#, Bastian Grewe3, Philipp Kirchner4, Arif Ekici4, Ana Beatriz 5 Oliveira Villela Silva1, Doris Jungnickl1, Markus Landthaler2,5, Armin Ensser1, and Klaus Überla1* 6 7 1 Institute of Clinical and Molecular Virology, University Hospital Erlangen, Friedrich-Alexander 8 Universität Erlangen-Nürnberg, Erlangen, Germany 9 2 Berlin Institute for Medical Systems Biology, Max-Delbrück-Center for Molecular Medicine in the 10 Helmholtz Association, Robert-Rössle-Strasse 10, 13125, Berlin, Germany 11 3 Department of Molecular and Medical Virology, Ruhr-University, Bochum, Germany 12 4 Institute of Human Genetics, University Hospital Erlangen, Friedrich-Alexander Universität Erlangen- 13 Nürnberg, Erlangen, Germany 14 5 IRI Life Sciences, Institute für Biologie, Humboldt Universität zu Berlin, Philippstraße 13, 10115, Berlin, 15 Germany 16 # these two authors contributed equally 17 18 19 *Corresponding author: 20 Prof. Dr. Klaus Überla 21 Institute of Clinical and Molecular Virology, University Hospital Erlangen 22 Friedrich-Alexander Universität Erlangen-Nürnberg 23 Schlossgarten 4, 91054 Erlangen 24 Germany 25 Tel: (+49) 9131-8523563 26 e-mail: [email protected] 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.02.04.934927; this version posted February 11, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. -

Análise Integrativa De Perfis Transcricionais De Pacientes Com
UNIVERSIDADE DE SÃO PAULO FACULDADE DE MEDICINA DE RIBEIRÃO PRETO PROGRAMA DE PÓS-GRADUAÇÃO EM GENÉTICA ADRIANE FEIJÓ EVANGELISTA Análise integrativa de perfis transcricionais de pacientes com diabetes mellitus tipo 1, tipo 2 e gestacional, comparando-os com manifestações demográficas, clínicas, laboratoriais, fisiopatológicas e terapêuticas Ribeirão Preto – 2012 ADRIANE FEIJÓ EVANGELISTA Análise integrativa de perfis transcricionais de pacientes com diabetes mellitus tipo 1, tipo 2 e gestacional, comparando-os com manifestações demográficas, clínicas, laboratoriais, fisiopatológicas e terapêuticas Tese apresentada à Faculdade de Medicina de Ribeirão Preto da Universidade de São Paulo para obtenção do título de Doutor em Ciências. Área de Concentração: Genética Orientador: Prof. Dr. Eduardo Antonio Donadi Co-orientador: Prof. Dr. Geraldo A. S. Passos Ribeirão Preto – 2012 AUTORIZO A REPRODUÇÃO E DIVULGAÇÃO TOTAL OU PARCIAL DESTE TRABALHO, POR QUALQUER MEIO CONVENCIONAL OU ELETRÔNICO, PARA FINS DE ESTUDO E PESQUISA, DESDE QUE CITADA A FONTE. FICHA CATALOGRÁFICA Evangelista, Adriane Feijó Análise integrativa de perfis transcricionais de pacientes com diabetes mellitus tipo 1, tipo 2 e gestacional, comparando-os com manifestações demográficas, clínicas, laboratoriais, fisiopatológicas e terapêuticas. Ribeirão Preto, 2012 192p. Tese de Doutorado apresentada à Faculdade de Medicina de Ribeirão Preto da Universidade de São Paulo. Área de Concentração: Genética. Orientador: Donadi, Eduardo Antonio Co-orientador: Passos, Geraldo A. 1. Expressão gênica – microarrays 2. Análise bioinformática por module maps 3. Diabetes mellitus tipo 1 4. Diabetes mellitus tipo 2 5. Diabetes mellitus gestacional FOLHA DE APROVAÇÃO ADRIANE FEIJÓ EVANGELISTA Análise integrativa de perfis transcricionais de pacientes com diabetes mellitus tipo 1, tipo 2 e gestacional, comparando-os com manifestações demográficas, clínicas, laboratoriais, fisiopatológicas e terapêuticas. -

Association of Gene Ontology Categories with Decay Rate for Hepg2 Experiments These Tables Show Details for All Gene Ontology Categories
Supplementary Table 1: Association of Gene Ontology Categories with Decay Rate for HepG2 Experiments These tables show details for all Gene Ontology categories. Inferences for manual classification scheme shown at the bottom. Those categories used in Figure 1A are highlighted in bold. Standard Deviations are shown in parentheses. P-values less than 1E-20 are indicated with a "0". Rate r (hour^-1) Half-life < 2hr. Decay % GO Number Category Name Probe Sets Group Non-Group Distribution p-value In-Group Non-Group Representation p-value GO:0006350 transcription 1523 0.221 (0.009) 0.127 (0.002) FASTER 0 13.1 (0.4) 4.5 (0.1) OVER 0 GO:0006351 transcription, DNA-dependent 1498 0.220 (0.009) 0.127 (0.002) FASTER 0 13.0 (0.4) 4.5 (0.1) OVER 0 GO:0006355 regulation of transcription, DNA-dependent 1163 0.230 (0.011) 0.128 (0.002) FASTER 5.00E-21 14.2 (0.5) 4.6 (0.1) OVER 0 GO:0006366 transcription from Pol II promoter 845 0.225 (0.012) 0.130 (0.002) FASTER 1.88E-14 13.0 (0.5) 4.8 (0.1) OVER 0 GO:0006139 nucleobase, nucleoside, nucleotide and nucleic acid metabolism3004 0.173 (0.006) 0.127 (0.002) FASTER 1.28E-12 8.4 (0.2) 4.5 (0.1) OVER 0 GO:0006357 regulation of transcription from Pol II promoter 487 0.231 (0.016) 0.132 (0.002) FASTER 6.05E-10 13.5 (0.6) 4.9 (0.1) OVER 0 GO:0008283 cell proliferation 625 0.189 (0.014) 0.132 (0.002) FASTER 1.95E-05 10.1 (0.6) 5.0 (0.1) OVER 1.50E-20 GO:0006513 monoubiquitination 36 0.305 (0.049) 0.134 (0.002) FASTER 2.69E-04 25.4 (4.4) 5.1 (0.1) OVER 2.04E-06 GO:0007050 cell cycle arrest 57 0.311 (0.054) 0.133 (0.002) -

Análise Correlacional Entre a Expressão Dos Fatores De Splicing E a Ocorrência De Splicing Alternativo Em Tecidos Humanos E De Camundongos
ANÁLISE CORRELACIONAL ENTRE A EXPRESSÃO DOS FATORES DE SPLICING E A OCORRÊNCIA DE SPLICING ALTERNATIVO EM TECIDOS HUMANOS E DE CAMUNDONGOS JULIO CÉSAR NUNES Dissertação apresentada à Fundação Antônio Prudente para a obtenção do título de Mestre em Ciências Área de Concentração: Oncologia Orientador: Dr. Sandro José de Souza São Paulo 2008 Livros Grátis http://www.livrosgratis.com.br Milhares de livros grátis para download. FICHA CATALOGRÁFICA Preparada pela Biblioteca da Fundação Antônio Prudente Nunes, Julio César Análise correlacional entre a expressão dos fatores de splicing e a ocorrência de splicing alternativo em tecidos humanos e de camundongos / Julio César Nunes – São Paulo, 2008. 79p. Dissertação (Mestrado) - Fundação Antônio Prudente. Curso de Pós-Graduação em Ciências - Área de concentração: Oncologia. Orientador: Sandro José Souza Descritores: 1. SPLICING ALTERNATIVO 2. BIOLOGIA MOLECULAR COMPUTACIONAL 3. CÂNCER 4. GENOMICA. AGRADECIMENTOS Agradeço à FAPESP e CAPES pela bolsa de Mestrado. Ao Sandro José de Souza agradeço toda orientação e conhecimento oferecido. Meus especiais agradecimentos ao Pedro Alexandre Favoretto Galante que dedicou atenção a minha formação no processo de Pós-Graduação na Fundação Antônio Prudente, bem como pela sua oficiosa co-orientação ao projeto de pesquisa. À grande família e amigos pela dedicação e incentivo a minha formação acadêmica. À Fundação Antônio Prudente, Hospital do Câncer e Instituto Ludwig de Pesquisa sobre o Câncer dedico os meus nobres agradecimentos finais. RESUMO Nunes JC. Análise correlacional entre a expressão dos fatores de splicing e a ocorrência de splicing alternativo em tecidos humanos e de camundongos. São Paulo; 2007. [Dissertacão de Mestrado - Fundação Antônio Prudente] Splicing alternativo desempenha uma significante função no aumento da complexidade genômica, produzindo um extenso número de mRNA e isoformas protéicas. -

Human Lectins, Their Carbohydrate Affinities and Where to Find Them
biomolecules Review Human Lectins, Their Carbohydrate Affinities and Where to Review HumanFind Them Lectins, Their Carbohydrate Affinities and Where to FindCláudia ThemD. Raposo 1,*, André B. Canelas 2 and M. Teresa Barros 1 1, 2 1 Cláudia D. Raposo * , Andr1 é LAQVB. Canelas‐Requimte,and Department M. Teresa of Chemistry, Barros NOVA School of Science and Technology, Universidade NOVA de Lisboa, 2829‐516 Caparica, Portugal; [email protected] 12 GlanbiaLAQV-Requimte,‐AgriChemWhey, Department Lisheen of Chemistry, Mine, Killoran, NOVA Moyne, School E41 of ScienceR622 Co. and Tipperary, Technology, Ireland; canelas‐ [email protected] NOVA de Lisboa, 2829-516 Caparica, Portugal; [email protected] 2* Correspondence:Glanbia-AgriChemWhey, [email protected]; Lisheen Mine, Tel.: Killoran, +351‐212948550 Moyne, E41 R622 Tipperary, Ireland; [email protected] * Correspondence: [email protected]; Tel.: +351-212948550 Abstract: Lectins are a class of proteins responsible for several biological roles such as cell‐cell in‐ Abstract:teractions,Lectins signaling are pathways, a class of and proteins several responsible innate immune for several responses biological against roles pathogens. such as Since cell-cell lec‐ interactions,tins are able signalingto bind to pathways, carbohydrates, and several they can innate be a immuneviable target responses for targeted against drug pathogens. delivery Since sys‐ lectinstems. In are fact, able several to bind lectins to carbohydrates, were approved they by canFood be and a viable Drug targetAdministration for targeted for drugthat purpose. delivery systems.Information In fact, about several specific lectins carbohydrate were approved recognition by Food by andlectin Drug receptors Administration was gathered for that herein, purpose. plus Informationthe specific organs about specific where those carbohydrate lectins can recognition be found by within lectin the receptors human was body. -

Network Effects of the Neuropsychiatric 15Q13.3 Microdeletion on the Transcriptome and Epigenome in Human Induced Neurons
bioRxiv preprint doi: https://doi.org/10.1101/772541; this version posted September 19, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Network effects of the neuropsychiatric 15q13.3 microdeletion on the transcriptome and epigenome in human induced neurons Siming Zhang (B.S., Stanford University), Xianglong Zhang (Ph.D., Stanford University), Shining Ma (Ph.D., Stanford University), Carolin Purmann (Ph.D., Stanford University), Kasey Davis (Ph.D., Stanford University), Wing Hung Wong (Ph.D., Stanford University), Jonathan Bernstein (MD, Ph.D., Stanford University), Joachim Hallmayer (MD, Stanford University), Alexander E Urban (Ph.D., Stanford University) Corresponding author: Alexander E Urban Address: 3165 Porter Drive, Stanford CA 94304 Phone: (650) 736-9528, Fax: (650) 725-4913 email: [email protected] bioRxiv preprint doi: https://doi.org/10.1101/772541; this version posted September 19, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Abstract Heterozygous deletions in the 15q13.3 region are associated with several neuropsychiatric disorders including autism, schizophrenia, and attention deficit hyperactivity disorder. Several genes within the 15q13.3 deletion region may play a role in neuronal dysfunction, based on association studies in humans and functional studies in mice, but the intermediate molecular mechanisms remain unknown. -

A Collection of Pre-Mrna Splicing Mutants in Arabidopsis Thaliana
G3: Genes|Genomes|Genetics Early Online, published on April 7, 2020 as doi:10.1534/g3.119.400998 1 1 A collection of pre-mRNA splicing mutants in Arabidopsis thaliana 2 3 4 Tatsuo Kanno1,a, Peter Venhuizen2,a, Ming-Tsung Wu3, Phebe Chiou1, Chia-Liang Chang1, 5 Maria Kalyna2,c, Antonius J.M. Matzke1,c and Marjori Matzke1,c 6 7 1Institute of Plant and Microbial Biology, Academia Sinica, 128, Sec. 2, Academia Rd., Nangang 8 District, Taipei, 11529 Taiwan 9 10 2Department of Applied Genetics and Cell Biology, University of Natural Resources and Life 11 Sciences - BOKU, Muthgasse 18, 1190 Vienna, Austria 12 13 3Department of Plant Sciences, University of Cambridge, Downing Street, CB2 3EA Cambridge, 14 UK 15 16 athese authors should be regarded as joint first authors 17 18 19 cCorresponding authors: 20 Antonius J.M. Matzke ([email protected]) 21 Tel: +886-2787-1135 22 Marjori Matzke ([email protected]) 23 Tel: +886-2787-1135 24 Maria Kalyna ([email protected]) 25 Tel: +43-1-47654-94112 26 27 28 29 30 31 32 © The Author(s) 2020. Published by the Genetics Society of America. 2 33 34 Running title: Arabidopsis pre-mRNA splicing mutants 35 36 Key words: Arabidopsis thaliana, CBP80. miRNAs, mutant screen, pre-mRNA splicing 37 38 39 40 Corresponding authors: 41 Antonius J.M. Matzke ([email protected]), Institute of Plant and Microbial 42 Biology, Academia Sinica, 128, Sec. 2, Academia Rd., Nangang District, Taipei, 11529 Taiwan 43 Tel: +886-2787-1135 44 45 Marjori Matzke ([email protected]), Institute of Plant and Microbial Biology, 46 Academia Sinica, 128, Sec. -

Systematic Analysis of Palatal Transcriptome to Identify Cleft Palate Genes Within Tgfβ3-Knockout Mice Alleles: RNA-Seq Analysis of Tgfβ3 Mice Ozturk Et Al
Systematic analysis of palatal transcriptome to identify cleft palate genes within TGFβ3-knockout mice alleles: RNA-Seq analysis of TGFβ3 Mice Ozturk et al. Ozturk et al. BMC Genomics 2013, 14:113 http://www.biomedcentral.com/1471-2164/14/113 Ozturk et al. BMC Genomics 2013, 14:113 http://www.biomedcentral.com/1471-2164/14/113 RESEARCH ARTICLE Open Access Systematic analysis of palatal transcriptome to identify cleft palate genes within TGFβ3-knockout mice alleles: RNA-Seq analysis of TGFβ3 Mice Ferhat Ozturk1,4, You Li2, Xiujuan Zhu1, Chittibabu Guda2,3 and Ali Nawshad1* Abstract Background: In humans, cleft palate (CP) accounts for one of the largest number of birth defects with a complex genetic and environmental etiology. TGFβ3 has been established as an important regulator of palatal fusion in mice and it has been shown that TGFβ3-null mice exhibit CP without any other major deformities. However, the genes that regulate cellular decisions and molecular mechanisms maintained by the TGFβ3 pathway throughout palatogenesis are predominantly unexplored. Our objective in this study was to analyze global transcriptome changes within the palate during different gestational ages within TGFβ3 knockout mice to identify TGFβ3-associated genes previously unknown to be associated with the development of cleft palate. We used deep sequencing technology, RNA-Seq, to analyze the transcriptome of TGFβ3 knockout mice at crucial stages of palatogenesis, including palatal growth (E14.5), adhesion (E15.5), and fusion (E16.5). Results: The overall transcriptome analysis of TGFβ3 wildtype mice (C57BL/6) reveals that almost 6000 genes were upregulated during the transition from E14.5 to E15.5 and more than 2000 were downregulated from E15.5 to E16.5. -

Article (Published Version)
Article The Candidate Schizophrenia Risk Gene DGCR2 Regulates Early Steps of Corticogenesis MOLINARD-CHENU, Aude, DAYER, Alexandre Abstract Alterations in early steps of cortical circuit assembly are thought to play a critical role in vulnerability to schizophrenia (SZ), but the pathogenic impact of SZ-risk mutations on corticogenesis remains to be determined. DiGeorge syndrome critical region 2 (DGCR2) is located in the 22q11.2 locus, whose deletion is a major risk factor for SZ. Moreover, exome sequencing of individuals with idiopathic SZ identified a rare missense mutation in DGCR2, further suggesting that DGCR2 is involved in SZ. Reference MOLINARD-CHENU, Aude, DAYER, Alexandre. The Candidate Schizophrenia Risk Gene DGCR2 Regulates Early Steps of Corticogenesis. Biological Psychiatry, 2018, vol. 83, no. 8, p. 692-706 DOI : 10.1016/j.biopsych.2017.11.015 PMID : 29305086 Available at: http://archive-ouverte.unige.ch/unige:124259 Disclaimer: layout of this document may differ from the published version. 1 / 1 Biological Psychiatry Archival Report The Candidate Schizophrenia Risk Gene DGCR2 Regulates Early Steps of Corticogenesis Aude Molinard-Chenu and Alexandre Dayer ABSTRACT BACKGROUND: Alterations in early steps of cortical circuit assembly are thought to play a critical role in vulnerability to schizophrenia (SZ), but the pathogenic impact of SZ-risk mutations on corticogenesis remains to be determined. DiGeorge syndrome critical region 2 (DGCR2) is located in the 22q11.2 locus, whose deletion is a major risk factor for SZ. Moreover, exome sequencing of individuals with idiopathic SZ identified a rare missense mutation in DGCR2, further suggesting that DGCR2 is involved in SZ. METHODS: Here we investigated the function of Dgcr2 and the pathogenic impact of the SZ-risk DGCR2 mutation in mouse corticogenesis using in utero electroporation targeted to projection neurons. -

Gene Ontology Functional Annotations and Pleiotropy
Network based analysis of genetic disease associations Sarah Gilman Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy under the Executive Committee of the Graduate School of Arts and Sciences COLUMBIA UNIVERSITY 2014 © 2013 Sarah Gilman All Rights Reserved ABSTRACT Network based analysis of genetic disease associations Sarah Gilman Despite extensive efforts and many promising early findings, genome-wide association studies have explained only a small fraction of the genetic factors contributing to common human diseases. There are many theories about where this “missing heritability” might lie, but increasingly the prevailing view is that common variants, the target of GWAS, are not solely responsible for susceptibility to common diseases and a substantial portion of human disease risk will be found among rare variants. Relatively new, such variants have not been subject to purifying selection, and therefore may be particularly pertinent for neuropsychiatric disorders and other diseases with greatly reduced fecundity. Recently, several researchers have made great progress towards uncovering the genetics behind autism and schizophrenia. By sequencing families, they have found hundreds of de novo variants occurring only in affected individuals, both large structural copy number variants and single nucleotide variants. Despite studying large cohorts there has been little recurrence among the genes implicated suggesting that many hundreds of genes may underlie these complex phenotypes. The question -

Detailed Characterization of Human Induced Pluripotent Stem Cells Manufactured for Therapeutic Applications
Stem Cell Rev and Rep DOI 10.1007/s12015-016-9662-8 Detailed Characterization of Human Induced Pluripotent Stem Cells Manufactured for Therapeutic Applications Behnam Ahmadian Baghbaderani 1 & Adhikarla Syama2 & Renuka Sivapatham3 & Ying Pei4 & Odity Mukherjee2 & Thomas Fellner1 & Xianmin Zeng3,4 & Mahendra S. Rao5,6 # The Author(s) 2016. This article is published with open access at Springerlink.com Abstract We have recently described manufacturing of hu- help determine which set of tests will be most useful in mon- man induced pluripotent stem cells (iPSC) master cell banks itoring the cells and establishing criteria for discarding a line. (MCB) generated by a clinically compliant process using cord blood as a starting material (Baghbaderani et al. in Stem Cell Keywords Induced pluripotent stem cells . Embryonic stem Reports, 5(4), 647–659, 2015). In this manuscript, we de- cells . Manufacturing . cGMP . Consent . Markers scribe the detailed characterization of the two iPSC clones generated using this process, including whole genome se- quencing (WGS), microarray, and comparative genomic hy- Introduction bridization (aCGH) single nucleotide polymorphism (SNP) analysis. We compare their profiles with a proposed calibra- Induced pluripotent stem cells (iPSCs) are akin to embryonic tion material and with a reporter subclone and lines made by a stem cells (ESC) [2] in their developmental potential, but dif- similar process from different donors. We believe that iPSCs fer from ESC in the starting cell used and the requirement of a are likely to be used to make multiple clinical products. We set of proteins to induce pluripotency [3]. Although function- further believe that the lines used as input material will be used ally identical, iPSCs may differ from ESC in subtle ways, at different sites and, given their immortal status, will be used including in their epigenetic profile, exposure to the environ- for many years or even decades.