A Collection of Pre-Mrna Splicing Mutants in Arabidopsis Thaliana
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
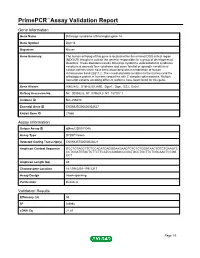
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name DiGeorge syndrome critical region gene 14 Gene Symbol Dgcr14 Organism Mouse Gene Summary The human ortholog of this gene is located within the minimal DGS critical region (MDGCR) thought to contain the gene(s) responsible for a group of developmental disorders. These disorders include DiGeorge syndrome velocardiofacial syndrome conotruncal anomaly face syndrome and some familial or sporadic conotruncal cardiac defects which have been associated with microdeletion of human chromosome band 22q11.2. The encoded protein localizes to the nucleus and the orthologous protein in humans co-purifies with C complex spliceosomes. Multiple transcript variants encoding different isoforms have been found for this gene. Gene Aliases AI462402, D16H22S1269E, Dgcr1, Dgsi, ES2, Es2el RefSeq Accession No. NC_000082.6, NT_039624.8, NT_187007.1 UniGene ID Mm.256480 Ensembl Gene ID ENSMUSG00000003527 Entrez Gene ID 27886 Assay Information Unique Assay ID qMmuCID0011048 Assay Type SYBR® Green Detected Coding Transcript(s) ENSMUST00000003621 Amplicon Context Sequence GCCTGTAGCTTCTCCACATCAGGGAAGAAGTCTCTCTGGATAACTGTCTGAAGTC CCTCGATGTACTCTTCTTCATCCAGGACCCGCTGCCTGCTTCTCGCAACTCCGG CCT Amplicon Length (bp) 82 Chromosome Location 16:17910201-17911217 Assay Design Intron-spanning Purification Desalted Validation Results Efficiency (%) 95 R2 0.9994 cDNA Cq 21.87 Page 1/5 PrimePCR™Assay Validation Report cDNA Tm (Celsius) 79.5 gDNA Cq 23.94 Specificity (%) 100 Information to assist with data interpretation is provided -

Multiple Cellular Proteins Interact with LEDGF/P75 Through a Conserved Unstructured Consensus Motif
ARTICLE Received 19 Jan 2015 | Accepted 1 Jul 2015 | Published 6 Aug 2015 DOI: 10.1038/ncomms8968 Multiple cellular proteins interact with LEDGF/p75 through a conserved unstructured consensus motif Petr Tesina1,2,3,*, Katerˇina Cˇerma´kova´4,*, Magdalena Horˇejsˇ´ı3, Katerˇina Procha´zkova´1, Milan Fa´bry3, Subhalakshmi Sharma4, Frauke Christ4, Jonas Demeulemeester4, Zeger Debyser4, Jan De Rijck4,**, Va´clav Veverka1,** & Pavlı´na Rˇeza´cˇova´1,3,** Lens epithelium-derived growth factor (LEDGF/p75) is an epigenetic reader and attractive therapeutic target involved in HIV integration and the development of mixed lineage leukaemia (MLL1) fusion-driven leukaemia. Besides HIV integrase and the MLL1-menin complex, LEDGF/p75 interacts with various cellular proteins via its integrase binding domain (IBD). Here we present structural characterization of IBD interactions with transcriptional repressor JPO2 and domesticated transposase PogZ, and show that the PogZ interaction is nearly identical to the interaction of LEDGF/p75 with MLL1. The interaction with the IBD is maintained by an intrinsically disordered IBD-binding motif (IBM) common to all known cellular partners of LEDGF/p75. In addition, based on IBM conservation, we identify and validate IWS1 as a novel LEDGF/p75 interaction partner. Our results also reveal how HIV integrase efficiently displaces cellular binding partners from LEDGF/p75. Finally, the similar binding modes of LEDGF/p75 interaction partners represent a new challenge for the development of selective interaction inhibitors. 1 Institute of Organic Chemistry and Biochemistry of the ASCR, v.v.i., Flemingovo nam. 2, 166 10 Prague, Czech Republic. 2 Department of Genetics and Microbiology, Faculty of Science, Charles University in Prague, Vinicna 5, 128 44 Prague, Czech Republic. -

1 Lessons from Non-Canonical Splicing 1 2 Christopher R Sibley1,2*, Lorea Blazquez1* and Jernej Ule1 3 4 Addresses: 5
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by UCL Discovery 1 Lessons from non-canonical splicing 2 3 Christopher R Sibley1,2*, Lorea Blazquez1* and Jernej Ule1 4 5 Addresses: 6 1Department of Molecular Neuroscience, UCL Institute of Neurology, Russell 7 Square House, Russell Square, London, WC1B 5EH, UK 8 9 2Division of Brain Sciences, Imperial College, Burlington Danes, DuCane Road, 10 London W12 0NN, UK 11 12 13 Additional information: 14 * These authors contributed equally to this work 15 16 17 Correspondence to JU 18 19 E-mail: [email protected] 20 21 22 DOI: 10.1038/nrgXXXX [office use only] 1 Abstract Recent improvements in experimental and computational techniques used to study the transcriptome have enabled an unprecedented view of RNA processing, revealing many previously unknown non-canonical splicing events. 5 This includes cryptic events located far from the currently annotated exons, and unconventional splicing mechanisms that have important roles in regulating gene expression. These non-canonical splicing events are a major source of newly emerging transcripts during evolution, especially when they involve sequences derived from transposable elements. They are therefore under precise 10 regulation and quality control, which minimises their potential to disrupt gene expression. While non-canonical splicing can lead to aberrant transcripts that cause many diseases, we also explain how it can be exploited for new therapeutic strategies. 15 Introduction The vast majority of human genes contain more than one exon, and therefore introns need to be spliced from the nascent transcript and exons joined to form an mRNA that can be translated into a protein. -

Splicing and Editing of Ionotropic Glutamate Receptors: a Comprehensive Analysis Based on Human RNA‑Seq Data
Cellular and Molecular Life Sciences (2021) 78:5605–5630 https://doi.org/10.1007/s00018-021-03865-z Cellular andMolecular Life Sciences ORIGINAL ARTICLE Splicing and editing of ionotropic glutamate receptors: a comprehensive analysis based on human RNA‑Seq data Robin Herbrechter1 · Nadine Hube1 · Raoul Buchholz1 · Andreas Reiner1 Received: 20 March 2021 / Revised: 12 May 2021 / Accepted: 22 May 2021 / Published online: 8 June 2021 © The Author(s) 2021 Abstract Ionotropic glutamate receptors (iGluRs) play key roles for signaling in the central nervous system. Alternative splicing and RNA editing are well-known mechanisms to increase iGluR diversity and to provide context-dependent regulation. Earlier work on isoform identifcation has focused on the analysis of cloned transcripts, mostly from rodents. We here set out to obtain a systematic overview of iGluR splicing and editing in human brain based on RNA-Seq data. Using data from two large-scale transcriptome studies, we established a workfow for the de novo identifcation and quantifcation of alternative splice and editing events. We detected all canonical iGluR splice junctions, assessed the abundance of alternative events described in the literature, and identifed new splice events in AMPA, kainate, delta, and NMDA receptor subunits. Notable events include an abundant transcript encoding the GluA4 amino-terminal domain, GluA4-ATD, a novel C-terminal GluD1 (delta receptor 1) isoform, GluD1-b, and potentially new GluK4 and GluN2C isoforms. C-terminal GluN1 splicing may be controlled by inclusion of a cassette exon, which shows preference for one of the two acceptor sites in the last exon. Moreo- ver, we identifed alternative untranslated regions (UTRs) and species-specifc diferences in splicing. -

CRNKL1 Is a Highly Selective Regulator of Intron-Retaining HIV-1 and Cellular Mrnas
bioRxiv preprint doi: https://doi.org/10.1101/2020.02.04.934927; this version posted February 11, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 CRNKL1 is a highly selective regulator of intron-retaining HIV-1 and cellular mRNAs 2 3 4 Han Xiao1, Emanuel Wyler2#, Miha Milek2#, Bastian Grewe3, Philipp Kirchner4, Arif Ekici4, Ana Beatriz 5 Oliveira Villela Silva1, Doris Jungnickl1, Markus Landthaler2,5, Armin Ensser1, and Klaus Überla1* 6 7 1 Institute of Clinical and Molecular Virology, University Hospital Erlangen, Friedrich-Alexander 8 Universität Erlangen-Nürnberg, Erlangen, Germany 9 2 Berlin Institute for Medical Systems Biology, Max-Delbrück-Center for Molecular Medicine in the 10 Helmholtz Association, Robert-Rössle-Strasse 10, 13125, Berlin, Germany 11 3 Department of Molecular and Medical Virology, Ruhr-University, Bochum, Germany 12 4 Institute of Human Genetics, University Hospital Erlangen, Friedrich-Alexander Universität Erlangen- 13 Nürnberg, Erlangen, Germany 14 5 IRI Life Sciences, Institute für Biologie, Humboldt Universität zu Berlin, Philippstraße 13, 10115, Berlin, 15 Germany 16 # these two authors contributed equally 17 18 19 *Corresponding author: 20 Prof. Dr. Klaus Überla 21 Institute of Clinical and Molecular Virology, University Hospital Erlangen 22 Friedrich-Alexander Universität Erlangen-Nürnberg 23 Schlossgarten 4, 91054 Erlangen 24 Germany 25 Tel: (+49) 9131-8523563 26 e-mail: [email protected] 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.02.04.934927; this version posted February 11, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. -

Análise Integrativa De Perfis Transcricionais De Pacientes Com
UNIVERSIDADE DE SÃO PAULO FACULDADE DE MEDICINA DE RIBEIRÃO PRETO PROGRAMA DE PÓS-GRADUAÇÃO EM GENÉTICA ADRIANE FEIJÓ EVANGELISTA Análise integrativa de perfis transcricionais de pacientes com diabetes mellitus tipo 1, tipo 2 e gestacional, comparando-os com manifestações demográficas, clínicas, laboratoriais, fisiopatológicas e terapêuticas Ribeirão Preto – 2012 ADRIANE FEIJÓ EVANGELISTA Análise integrativa de perfis transcricionais de pacientes com diabetes mellitus tipo 1, tipo 2 e gestacional, comparando-os com manifestações demográficas, clínicas, laboratoriais, fisiopatológicas e terapêuticas Tese apresentada à Faculdade de Medicina de Ribeirão Preto da Universidade de São Paulo para obtenção do título de Doutor em Ciências. Área de Concentração: Genética Orientador: Prof. Dr. Eduardo Antonio Donadi Co-orientador: Prof. Dr. Geraldo A. S. Passos Ribeirão Preto – 2012 AUTORIZO A REPRODUÇÃO E DIVULGAÇÃO TOTAL OU PARCIAL DESTE TRABALHO, POR QUALQUER MEIO CONVENCIONAL OU ELETRÔNICO, PARA FINS DE ESTUDO E PESQUISA, DESDE QUE CITADA A FONTE. FICHA CATALOGRÁFICA Evangelista, Adriane Feijó Análise integrativa de perfis transcricionais de pacientes com diabetes mellitus tipo 1, tipo 2 e gestacional, comparando-os com manifestações demográficas, clínicas, laboratoriais, fisiopatológicas e terapêuticas. Ribeirão Preto, 2012 192p. Tese de Doutorado apresentada à Faculdade de Medicina de Ribeirão Preto da Universidade de São Paulo. Área de Concentração: Genética. Orientador: Donadi, Eduardo Antonio Co-orientador: Passos, Geraldo A. 1. Expressão gênica – microarrays 2. Análise bioinformática por module maps 3. Diabetes mellitus tipo 1 4. Diabetes mellitus tipo 2 5. Diabetes mellitus gestacional FOLHA DE APROVAÇÃO ADRIANE FEIJÓ EVANGELISTA Análise integrativa de perfis transcricionais de pacientes com diabetes mellitus tipo 1, tipo 2 e gestacional, comparando-os com manifestações demográficas, clínicas, laboratoriais, fisiopatológicas e terapêuticas. -

Análise Correlacional Entre a Expressão Dos Fatores De Splicing E a Ocorrência De Splicing Alternativo Em Tecidos Humanos E De Camundongos
ANÁLISE CORRELACIONAL ENTRE A EXPRESSÃO DOS FATORES DE SPLICING E A OCORRÊNCIA DE SPLICING ALTERNATIVO EM TECIDOS HUMANOS E DE CAMUNDONGOS JULIO CÉSAR NUNES Dissertação apresentada à Fundação Antônio Prudente para a obtenção do título de Mestre em Ciências Área de Concentração: Oncologia Orientador: Dr. Sandro José de Souza São Paulo 2008 Livros Grátis http://www.livrosgratis.com.br Milhares de livros grátis para download. FICHA CATALOGRÁFICA Preparada pela Biblioteca da Fundação Antônio Prudente Nunes, Julio César Análise correlacional entre a expressão dos fatores de splicing e a ocorrência de splicing alternativo em tecidos humanos e de camundongos / Julio César Nunes – São Paulo, 2008. 79p. Dissertação (Mestrado) - Fundação Antônio Prudente. Curso de Pós-Graduação em Ciências - Área de concentração: Oncologia. Orientador: Sandro José Souza Descritores: 1. SPLICING ALTERNATIVO 2. BIOLOGIA MOLECULAR COMPUTACIONAL 3. CÂNCER 4. GENOMICA. AGRADECIMENTOS Agradeço à FAPESP e CAPES pela bolsa de Mestrado. Ao Sandro José de Souza agradeço toda orientação e conhecimento oferecido. Meus especiais agradecimentos ao Pedro Alexandre Favoretto Galante que dedicou atenção a minha formação no processo de Pós-Graduação na Fundação Antônio Prudente, bem como pela sua oficiosa co-orientação ao projeto de pesquisa. À grande família e amigos pela dedicação e incentivo a minha formação acadêmica. À Fundação Antônio Prudente, Hospital do Câncer e Instituto Ludwig de Pesquisa sobre o Câncer dedico os meus nobres agradecimentos finais. RESUMO Nunes JC. Análise correlacional entre a expressão dos fatores de splicing e a ocorrência de splicing alternativo em tecidos humanos e de camundongos. São Paulo; 2007. [Dissertacão de Mestrado - Fundação Antônio Prudente] Splicing alternativo desempenha uma significante função no aumento da complexidade genômica, produzindo um extenso número de mRNA e isoformas protéicas. -

The Function and Evolution of C2H2 Zinc Finger Proteins and Transposons
The function and evolution of C2H2 zinc finger proteins and transposons by Laura Francesca Campitelli A thesis submitted in conformity with the requirements for the degree of Doctor of Philosophy Department of Molecular Genetics University of Toronto © Copyright by Laura Francesca Campitelli 2020 The function and evolution of C2H2 zinc finger proteins and transposons Laura Francesca Campitelli Doctor of Philosophy Department of Molecular Genetics University of Toronto 2020 Abstract Transcription factors (TFs) confer specificity to transcriptional regulation by binding specific DNA sequences and ultimately affecting the ability of RNA polymerase to transcribe a locus. The C2H2 zinc finger proteins (C2H2 ZFPs) are a TF class with the unique ability to diversify their DNA-binding specificities in a short evolutionary time. C2H2 ZFPs comprise the largest class of TFs in Mammalian genomes, including nearly half of all Human TFs (747/1,639). Positive selection on the DNA-binding specificities of C2H2 ZFPs is explained by an evolutionary arms race with endogenous retroelements (EREs; copy-and-paste transposable elements), where the C2H2 ZFPs containing a KRAB repressor domain (KZFPs; 344/747 Human C2H2 ZFPs) are thought to diversify to bind new EREs and repress deleterious transposition events. However, evidence of the gain and loss of KZFP binding sites on the ERE sequence is sparse due to poor resolution of ERE sequence evolution, despite the recent publication of binding preferences for 242/344 Human KZFPs. The goal of my doctoral work has been to characterize the Human C2H2 ZFPs, with specific interest in their evolutionary history, functional diversity, and coevolution with LINE EREs. -

DGCR6 at the Proximal Part of the Digeorge Critical Region Is Involved in Conotruncal Heart Defects
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Institutional Repository : the EHIME area OPEN Citation: Human Genome Variation (2015) 2, 15004; doi:10.1038/hgv.2015.4 © 2015 The Japan Society of Human Genetics All rights reserved 2054-345X/15 www.nature.com/hgv ARTICLE DGCR6 at the proximal part of the DiGeorge critical region is involved in conotruncal heart defects Wenming Gao1, Takashi Higaki1, Minenori Eguchi-Ishimae1, Hidehiko Iwabuki1, Zhouying Wu1, Eiichi Yamamoto2, Hidemi Takata1, Masaaki Ohta1, Issei Imoto3, Eiichi Ishii1 and Mariko Eguchi1 Cardiac anomaly is one of the hallmarks of DiGeorge syndrome (DGS), observed in approximately 80% of patients. It often shows a characteristic morphology, termed as conotruncal heart defects. In many cases showing only the conotruncal heart defect, deletion of 22q11.2 region cannot be detected by fluorescence in situ hybridization (FISH), which is used to detect deletion in DGS. We investigated the presence of genomic aberrations in six patients with congenital conotruncal heart defects, who show no deletion at 22q11.2 in an initial screening by FISH. In these patients, no abnormalities were identified in the coding region of the TBX1 gene, one of the key genes responsible for the phenotype of DGS. However, when copy number alteration was analyzed by high-resolution array analysis, a small deletion or duplication in the proximal end of DiGeorge critical region was detected in two patients. The affected region contains the DGCR6 and PRODH genes. DGCR6 has been reported to affect the expression of the TBX1 gene. Our results suggest that altered dosage of gene(s) other than TBX1, possibly DGCR6, may also be responsible for the development of conotruncal heart defects observed in patients with DGS and, in particular, in those with stand-alone conotruncal heart defects. -

IWS1 Antibody A
Revision 1 C 0 2 - t IWS1 Antibody a e r o t S Orders: 877-616-CELL (2355) [email protected] Support: 877-678-TECH (8324) 1 8 Web: [email protected] 6 www.cellsignal.com 5 # 3 Trask Lane Danvers Massachusetts 01923 USA For Research Use Only. Not For Use In Diagnostic Procedures. Applications: Reactivity: Sensitivity: MW (kDa): Source: UniProt ID: Entrez-Gene Id: WB, IP, IF-IC H M R Endogenous 140 Rabbit Q96ST2 55677 Product Usage Information Application Dilution Western Blotting 1:1000 Immunoprecipitation 1:50 Immunofluorescence (Immunocytochemistry) 1:200 Storage Supplied in 10 mM sodium HEPES (pH 7.5), 150 mM NaCl, 100 µg/ml BSA and 50% glycerol. Store at –20°C. Do not aliquot the antibody. Specificity / Sensitivity IWS1 Antibody detects endogenous levels of total IWS1 protein. Species Reactivity: Human, Mouse, Rat Source / Purification Polyclonal antibodies are produced by immunizing animals with a synthetic peptide corresponding to residues near the amino terminus of human IWS1 protein. Antibodies are purified by protein A and peptide affinity chromatography. Background Various steps in gene expression, such as mRNA processing, surveillance, export, and synthesis are coupled to transcription elongation (1,2). The C-terminal domain (CTD) of the large subunit of RNA polymerase II plays an important role in the integration of these different steps (1,2). IWS1 interacts with Spt6, a CTD-binding transcription elongation factor and H3 chaperone (1,2). IWS1 also recruits another CTD-binding protein, HYPB/Setd2 histone methyltransferase, to the RNA polymerase II complex for elongation-coupled H3K36 trimethylation (2). Thus, IWS1 links Spt6 and HYPB/Setd2 in a large complex and regulates mRNA synthesis and histone methylation at the co- transcriptional level (2). -

Dynamics of Transcription-Dependent H3k36me3 Marking by the SETD2:IWS1:SPT6 Ternary Complex
bioRxiv preprint doi: https://doi.org/10.1101/636084; this version posted May 14, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Dynamics of transcription-dependent H3K36me3 marking by the SETD2:IWS1:SPT6 ternary complex Katerina Cermakova1, Eric A. Smith1, Vaclav Veverka2, H. Courtney Hodges1,3,4,* 1 Department of Molecular & Cellular Biology, Center for Precision Environmental Health, and Dan L Duncan Comprehensive Cancer Center, Baylor College of Medicine, Houston, TX, 77030, USA 2 Institute of Organic Chemistry and Biochemistry, Czech Academy of Sciences, Prague, Czech Republic 3 Center for Cancer Epigenetics, The University of Texas MD Anderson Cancer Center, Houston, TX, 77030, USA 4 Department of Bioengineering, Rice University, Houston, TX, 77005, USA * Lead contact; Correspondence to: [email protected] Abstract The genome-wide distribution of H3K36me3 is maintained SETD2 contributes to gene expression by marking gene through various mechanisms. In human cells, H3K36 is bodies with H3K36me3, which is thought to assist in the mono- and di-methylated by eight distinct histone concentration of transcription machinery at the small portion methyltransferases; however, the predominant writer of the of the coding genome. Despite extensive genome-wide data trimethyl mark on H3K36 is SETD21,11,12. Interestingly, revealing the precise localization of H3K36me3 over gene SETD2 is a major tumor suppressor in clear cell renal cell bodies, the physical basis for the accumulation, carcinoma13, breast cancer14, bladder cancer15, and acute maintenance, and sharp borders of H3K36me3 over these lymphoblastic leukemias16–18. In these settings, mutations sites remains rudimentary. -

A Master Autoantigen-Ome Links Alternative Splicing, Female Predilection, and COVID-19 to Autoimmune Diseases
bioRxiv preprint doi: https://doi.org/10.1101/2021.07.30.454526; this version posted August 4, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. A Master Autoantigen-ome Links Alternative Splicing, Female Predilection, and COVID-19 to Autoimmune Diseases Julia Y. Wang1*, Michael W. Roehrl1, Victor B. Roehrl1, and Michael H. Roehrl2* 1 Curandis, New York, USA 2 Department of Pathology, Memorial Sloan Kettering Cancer Center, New York, USA * Correspondence: [email protected] or [email protected] 1 bioRxiv preprint doi: https://doi.org/10.1101/2021.07.30.454526; this version posted August 4, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Abstract Chronic and debilitating autoimmune sequelae pose a grave concern for the post-COVID-19 pandemic era. Based on our discovery that the glycosaminoglycan dermatan sulfate (DS) displays peculiar affinity to apoptotic cells and autoantigens (autoAgs) and that DS-autoAg complexes cooperatively stimulate autoreactive B1 cell responses, we compiled a database of 751 candidate autoAgs from six human cell types. At least 657 of these have been found to be affected by SARS-CoV-2 infection based on currently available multi-omic COVID data, and at least 400 are confirmed targets of autoantibodies in a wide array of autoimmune diseases and cancer.