1 1 Graphical Abstract
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Transport of Dangerous Goods
ST/SG/AC.10/1/Rev.16 (Vol.I) Recommendations on the TRANSPORT OF DANGEROUS GOODS Model Regulations Volume I Sixteenth revised edition UNITED NATIONS New York and Geneva, 2009 NOTE The designations employed and the presentation of the material in this publication do not imply the expression of any opinion whatsoever on the part of the Secretariat of the United Nations concerning the legal status of any country, territory, city or area, or of its authorities, or concerning the delimitation of its frontiers or boundaries. ST/SG/AC.10/1/Rev.16 (Vol.I) Copyright © United Nations, 2009 All rights reserved. No part of this publication may, for sales purposes, be reproduced, stored in a retrieval system or transmitted in any form or by any means, electronic, electrostatic, magnetic tape, mechanical, photocopying or otherwise, without prior permission in writing from the United Nations. UNITED NATIONS Sales No. E.09.VIII.2 ISBN 978-92-1-139136-7 (complete set of two volumes) ISSN 1014-5753 Volumes I and II not to be sold separately FOREWORD The Recommendations on the Transport of Dangerous Goods are addressed to governments and to the international organizations concerned with safety in the transport of dangerous goods. The first version, prepared by the United Nations Economic and Social Council's Committee of Experts on the Transport of Dangerous Goods, was published in 1956 (ST/ECA/43-E/CN.2/170). In response to developments in technology and the changing needs of users, they have been regularly amended and updated at succeeding sessions of the Committee of Experts pursuant to Resolution 645 G (XXIII) of 26 April 1957 of the Economic and Social Council and subsequent resolutions. -
![United States Patent (19) [11] 3,932,402 Norell (45) Jan](https://docslib.b-cdn.net/cover/6379/united-states-patent-19-11-3-932-402-norell-45-jan-626379.webp)
United States Patent (19) [11] 3,932,402 Norell (45) Jan
United States Patent (19) [11] 3,932,402 Norell (45) Jan. 13, 1976 54 PREPARATION OF SYM-TRAZINES BY 3,071,586 111963 Sandner et al...................... 260/248 TRIMERIZATION OF NITRLES IN 3,095,414 6/1963 Spain hour........................... 260/248 3,238,138 3/1966 Braunwarth et al... ... 260/248 X TRIFLUOROMETHANESULFONIC ACD 3,654, 192 4/1972 Vogel.................................. 260/2 R 75 Inventor: John R. Norell, Bartlesville, Okla. OTHER PUBLICATIONS 73 Assignee: Phillips Petroleum Company, Cool et al., J. Chem. Soc., 1941, pp. 278-282. Bartlesville, Okla. Elderfield, “Heterocyclic Compounds", Vol. 7, Ch. 7, pp. 642, 644 and 686. 22) Filed: June 24, 1974 Migrdichian, "The Chemistry of Organic Cyanogen 21 ) Appl. No.: 482,257 Compounds,' Ch. 17, pp. 356, 357 and 367. (52) U.S. Cl............................................ 260/248 CS Primary Examiner-John M. Ford 51) Int. Cl’........................................ C07D 251/24 58 Field of Search............................... 260/248 CS 57 ABSTRACT Aliphatic and aromatic nitriles are trimerized to sub 56) References Cited stituted symmetrical triazines in the presence of UNITED STATES PATENTS trifluoromethanesulfonic acid. 3,060, 179 lof 1962 Toland................................ 260/248 16 Claims, No Drawings 3,932,402 2 In one embodiment of this invention, the organic PREPARATION OF SYM-TRAZINES BY nitrile is an aliphatic nitrile. The product of this process TRIMERIZATION OF NITRILES IN is a 2,4,6-trialkyl-1,3,5-triazine. TRFLUOROMETHANESULFONIC ACID In another embodiment of this invention, the organic This invention relates to substituted sym-triazines. In 5 nitrile is an aromatic or substituted aromatic nitrile in one aspect it relates to the preparation of alkyl-sub which case the product of the process is a 2,4,6-triaryl stituted sym-triazines. -

Natural Product Isolation – How to Get from Biological Material to Pure Compounds Cite This: Nat
NPR View Article Online REVIEW View Journal | View Issue Natural product isolation – how to get from biological material to pure compounds Cite this: Nat. Prod. Rep., 2013, 30, 525 Franz Bucar,*a Abraham Wubea and Martin Schmidb Covering: 2008 to 2012 Since the last comprehensive review by Otto Sticher on natural product isolation in NPR (O. Sticher, Nat. Prod. Rep., 2008, 25, 517), a plethora of new reports on isolation of secondary compounds from higher plants, marine organisms and microorganisms has been published. Although methods described earlier like the liquid-solid chromatographic techniques (VLC, FC, MPLC, HPLC) or partition chromatographic methods are still the major tools for isolating pure compounds, some developments like hydrophilic interaction chromatography (HILIC) have not been fully covered in previous reviews. Furthermore, examples of using different preparative solid-phase extraction (SPE) columns including molecular imprinting technology have been included. Special attention is given to chiral stationary phases in fi Received 25th October 2012 isolation of natural products. Methods for proper identi cation of plant material, problems of post- harvest changes in plant material, extraction methods including application of ionic liquids, de- DOI: 10.1039/c3np20106f replication procedures during natural product isolation are further issues to be discussed by the review. www.rsc.org/npr Selected work published between 2008 and mid-2012 is covered. 1 Introduction 4.2 Column chromatographic methods 2 Authentication and preparation of plant material/ 4.2.1 Vacuum liquid chromatography (VLC) marine organisms 4.2.2 Flash chromatography (FC) 2.1 Morphological/anatomical analysis 4.2.3 Low-pressure liquid chromatography (LPLC) 2.2 TLC/HPTLC analysis 4.2.4 Medium-pressure liquid chromatography (MPLC) Open Access Article. -

Safety Data Sheet
1/7 Isobutyronitrile,1181E-2,28/07/2020 Date of issue: 05/02/2019 Date of revision: 28/07/2020 Safety Data Sheet 1. Identification of the substance/mixture and of the company/undertaking Product identifier: Product name: Isobutyronitrile SDS No. : 1181E-2 Details of the supplier of the safety data sheet Manufacturer/Supplier: KISHIDA CHEMICAL CO., LTD. Address: 3-1, Honmachibashi, Chuo-ku,Osaka ,JAPAN Division: Safety Management Dept. of Chemicals Telephone number: +81-6-6946-8061 FAX: +81-6-6946-1607 e-mail address: [email protected] 2. Hazards identification GHS classification and label elements of the product Classification of the substance or mixture PHYSICAL AND CHEMICAL HAZARDS Flammable liquids: Category 2 HEALTH HAZARDS Acute toxicity (Oral): Category 3 Acute toxicity (Dermal): Category 2 Acute toxicity (Inhalation): Category 3 Serious eye damage/eye irritation: Category 2 Specific target organ toxicity - single exposure: Category 2(liver) Specific target organ toxicity - single exposure: Category 3 (Respiratory tract irritation) (Note) GHS classification without description: Not classified/Classification not possible Label elements Signal word: Danger HAZARD STATEMENT Highly flammable liquid and vapor Toxic if swallowed Fatal in contact with skin Toxic if inhaled Causes serious eye irritation May cause damage to organs after single exposure(liver) May cause respiratory irritation PRECAUTIONARY STATEMENT Prevention Keep away from heat/sparks/open flames/hot surfaces. - No smoking. Keep container tightly closed. Ground/bond container and receiving equipment. Use explosion-proof electrical/ventilating/lighting equipment. Use only non-sparking tools. Take precautionary measures against static discharge. Do not breathe dust/fume/gas/mist/vapors/spray. -

1 Titan's Organic Aerosols: Molecular Composition And
TITAN’S ORGANIC AEROSOLS: MOLECULAR COMPOSITION AND STRUCTURE OF LABORATORY ANALOGUES INFERRED FROM PYROLYSIS GAS CHROMATOGRAPHY MASS SPECTROMETRY ANALYSIS Marietta Morissona,b,*, Cyril Szopab,c, NatHalie Carrascob,c, Arnaud BucHa and Thomas Gautierd a LGPM Laboratoire de Génie des Procédés et Matériaux, Ecole CentraleSupelec, Châtenay-Malabry Cedex, France b LATMOS/IPSL, UVSQ Université Paris-Saclay, UPMC Univ. Paris 06, CNRS, Guyancourt, France c Institut Universitaire de France, Paris F-75005, France d NASA Goddard Space FligHt Center, Greenbelt, MD, United States Abstract Analogues of Titan’s aerosols are of primary interest in the understanding of Titan’s atmospheric chemistry and climate, and in tHe development of in situ instrumentation for future space missions. Numerous studies have been carried out to characterize laboratory analogues of Titan aerosols (tholins), but their molecular composition and structure are still poorly known. If pyrolysis gas chromatograpHy mass spectrometry (pyr-GCMS) Has been used for years to give clues about their chemical composition, HigHly disparate results were obtained witH tHis tecHnique. THey can be attributed to tHe variety of analytical conditions used for pyr-GCMS analyses, and/or to differences in the nature of the analogues analyzed, tHat were produced witH different laboratory set-ups under various operating conditions. In order to Have a better description of Titan’s tHolin’s molecular composition by pyr-GCMS, we carried out a systematic study witH two major objectives: (i) exploring tHe pyr-GCMS analytical parameters to find the optimal ones for the detection of a wide range of chemical products allowing a cHaracterization of tHe tHolins composition as comprehensive as possible, and (ii) HigHligHting tHe role of the CH4 ratio in tHe gaseous reactive medium on the tholin’s molecular structure. -
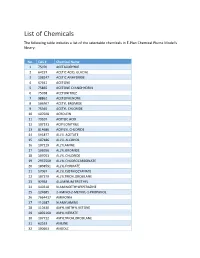
List of Chemicals the Following Table Includes a List of the Selectable Chemicals in E‐Plan Chemical Plume Model's Library
List of Chemicals The following table includes a list of the selectable chemicals in E‐Plan Chemical Plume Model's library: No. CAS # Chemical Name 1 75070 ACETALDEHYDE 2 64197 ACETIC ACID, GLACIAL 3 108247 ACETIC ANHYDRIDE 4 67641 ACETONE 5 75865 ACETONE CYANOHYDRIN 6 75058 ACETONITRILE 7 98862 ACETOPHENONE 8 506967 ACETYL BROMIDE 9 75365 ACETYL CHLORIDE 10 107028 ACROLEIN 11 79107 ACRYLIC ACID 12 107131 ACRYLONITRILE 13 814686 ACRYLYL CHLORIDE 14 591877 ALLYL ACETATE 15 107186 ALLYL ALCOHOL 16 107119 ALLYLAMINE 17 106956 ALLYL BROMIDE 18 107051 ALLYL CHLORIDE 19 2937500 ALLYL CHLOROCARBONATE 20 1838591 ALLYL FORMATE 21 57067 ALLYL ISOTHIOCYANATE 22 107379 ALLYLTRICHLOROSILANE 23 97938 ALUMINUM TRIETHYL 24 140318 N‐AMINOETHYLPIPERAZINE 25 124685 2‐AMINO‐2‐METHYL‐1‐PROPANOL 26 7664417 AMMONIA 27 110587 N‐AMYLAMINE 28 110430 AMYL METHYL KETONE 29 1002160 AMYL NITRATE 30 107722 AMYLTRICHLOROSILANE 31 62533 ANILINE 32 100663 ANISOLE No. CAS # Chemical Name 33 7647189 ANTIMONY PENTACHLORIDE 34 7783702 ANTIMONY PENTAFLUORIDE 35 7784341 ARSENIC TRICHLORIDE 36 7784421 ARSINE 37 71432 BENZENE 38 100470 BENZONITRILE 39 98077 BENZOTRICHLORIDE 40 98088 BENZOTRIFLUORIDE 41 98884 BENZOYL CHLORIDE 42 100469 BENZYLAMINE 43 100390 BENZYL BROMIDE 44 100447 BENZYL CHLORIDE 45 501531 BENZYL CHLOROFORMATE 46 98873 BENZYLIDENE CHLORIDE 47 10294334 BORON TRIBROMIDE 48 10294345 BORON TRICHLORIDE 49 7637072 BORON TRIFLUORIDE 50 109637 BORON TRIFLUORIDE DIETHYL ETHERATE 51 353424 BORON TRIFLUORIDE DIMETHYL ETHERATE 52 7726956 BROMINE 53 13863417 BROMINE CHLORIDE 54 7789302 BROMINE PENTAFLUORIDE 55 7787715 BROMINE TRIFLUORIDE 56 598312 BROMOACETONE 57 108861 BROMOBENZENE 58 109659 1‐BROMOBUTANE 59 353593 BROMOCHLORODIFLUOROMETHANE 60 74975 BROMOCHLOROMETHANE 61 540512 2‐BROMOETHANOL 62 75252 BROMOFORM 63 107824 1‐BROMO‐3‐METHYLBUTANE 64 75638 BROMOTRIFLUOROMETHANE 65 106990 1,3‐BUTADIENE 66 106978 BUTANE 67 106989 1‐BUTENE 68 590181 2‐BUTENE‐CIS 69 624646 2‐BUTENE‐TRANS 70 123864 BUTYL ACETATE 71 105464 SEC‐BUTYL ACETATE No. -

Synthesis of Fluorinated Amino Acids and Their Derivatives in Flow
Synthesis of Fluorinated Amino Acids and Their Derivatives in Flow Inaugural-Dissertation to obtain the academic degree Doctor rerum naturalium (Dr. rer. nat) submitted to the Department of Biology, Chemistry and Pharmacy of Freie Universität Berlin by Stella Vukelić From Zagreb, Croatia July, 2016 ______________________________________________________________________________________________________________________________________________________________________________________ ______________________________________________________________________________________________________________________________________________________________________________________ This work was performed between October 2012 and April 2016 under the guidance of Prof. Dr. Beate Koksch and Prof. Dr. Peter H. Seeberger in the Department of Biomolecular Systems, Max Planck Institute of Colloids and Interfaces Potsdam and the Institute of Chemistry and Biochemistry, Freie Universität Berlin. I hereby declare that this PhD thesis was prepared autonomously. Content, quotes or images previously published by third parties are indicated by referring to the original work. The work presented in this dissertation resulted in the following publications: • Stella Vukelić, Kerry Gilmore, Beate Koksch, Peter H. Seeberger, “A Sustainable Semi- Continuous Flow Synthesis of Hydantoins”, Eur. J. Of Chem. A, 2016, DOI: 10.1002/chem.201602609. • Stella Vukelić, Johann Moschner, Susanne Huhmann, Rita Fernandes, Allison Ann Berger, Beate Koksch, “Synthesis of side-chain fluorinated -

Reaction Pathways in the Chlorination of Amino Acids
Department of Chemistry Reaction Pathways in the Chlorination of Amino Acids Zuo Tong How This thesis is presented for the Degree of Doctor of Philosophy Of Curtin University June 2016 Declaration To the best of my knowledge and belief this thesis contains no material previously published by any other person except where due acknowledgment has been made. This thesis contains no material which has been accepted for the award of any other degree or diploma in any university. Signature: ____________ Zuo Tong How Date: 30/06/2016 Acknowledgments I am very grateful to many individuals for the help and support that I’ve received over the past years. Especially to the following people: My Principal Supervisor, A/Prof. Cynthia Joll, for her continual guidance and insights for the project; for her support and encouragement; and for her thorough review of this Thesis. My Co-Supervisor, Dr. Kathryn Linge, for her guidance and encouragement throughout my PhD program; for her advice on experimental planning and design; and her insightful feedback on this Thesis. My Co-Supervisor, Dr. Francesco Busetti, for his helpful insight and technical advice on LC-MS; for his contribution to experimental planning and design; and his helpful feedback on the Thesis. Mr. Geoff Chidlow, for his invaluable technical assistance and advice on the operation of GC-MS instrumentation. The staff and my fellow students in the Department of Chemistry, past and present, for their support, assistance and companionship over the years, particularly Ina Kristiana, Deborah Liew, Jace Tan, Mark Maric, Patrick Fritz, Krishnan Murugappan and Rhys Tilbury. The staff of the Water Corporation of Western Australia, for assistance in the provision of samples and operational information for my project. -

Aliphatic Nitriles Final AEGL Document
VOLUME 16 Committee on Acute Exposure Guideline Levels Committee on Toxicology Board on Environmental Studies and Toxicology Division on Earth and Life Studies THE NATIONAL ACADEMIES PRESS 500 FIFTH STREET, NW WASHINGTON, DC 20001 NOTICE: The project that is the subject of this report was approved by the Governing Board of the National Research Council, whose members are drawn from the councils of the National Academy of Sciences, the National Academy of Engineering, and the Institute of Medicine. The members of the committee responsible for the report were chosen for their special competences and with regard for appropriate balance. This project was supported by Contract No. W81K04-11-D-0017 and EP-W-09-007 between the National Academy of Sciences and the U.S. Department of Defense and the U.S. Environmental Protection Agency. Any opinions, findings, conclusions, or recommendations expressed in this publication are those of the author(s) and do not necessarily reflect the view of the organizations or agencies that provided support for this project. International Standard Book Number-13: 978-0-309-30096-4 International Standard Book Number-10: 0-309-30096-7 Additional copies of this report are available for sale from the National Academies Press, 500 Fifth Street, N.W., Keck 360, Washington, DC 20001; (800) 624-6242 or (202) 3343313; Internet, http://www.nap.edu/. Copyright 2014 by the National Academy of Sciences. All rights reserved. Printed in the United States of America The National Academy of Sciences is a private, nonprofit, self-perpetuating society of distinguished scholars engaged in scientific and engineering research, dedicated to the furtherance of science and technology and to their use for the general welfare. -

The NIOSH Occupational Exposure Banding Process for Chemical Risk Management
TECHNICAL REPORT The NIOSH Occupational Exposure Banding Process for Chemical Risk Management Centers for Disease Control and Prevention National Institute for Occupational Safety and Health This page intentionally blank. Technical Report The NIOSH Occupational Exposure Banding Process for Chemical Risk Management DEPARTMENT OF HEALTH AND HUMAN SERVICES Centers for Disease Control and Prevention National Institute for Occupational Safety and Health This document is in the public domain and may be freely copied or reprinted. Disclaimer Mention of any company or product does not constitute endorsement by the National Institute for Occupational Safety and Health, Centers for Disease Control and Prevention. In addition, citations to websites external to NIOSH do not constitute NIOSH endorsement of the sponsor- ing organizations or their programs or products. Furthermore, NIOSH is not responsible for the content of these websites. All web addresses referenced in this document were accessible as of the publication date. Get More Information Find NIOSH products and get answers to workplace safety and health questions: 1-800-CDC-INFO (1-800-232-4636) | TTY: 1-888-232-6348 CDC/NIOSH INFO: cdc.gov/info | cdc.gov/niosh Monthly NIOSH eNews: https://www.cdc.gov/niosh/enews/ Suggested Citation NIOSH [2019]. Technical report: The NIOSH occupational exposure banding process for chemical risk management. By Lentz TJ, Seaton M, Rane P, Gilbert SJ, McKernan LT, Whit- taker C. Cincinnati, OH: U.S. Department of Health and Human Services, Centers for Dis- ease Control and Prevention, National Institute for Occupational Safety and Health, DHHS (NIOSH) Publication No. 2019-132, https://doi.org/10.26616/NIOSHPUB2019132. -

Nitrile Gas Chemistry in Titan Atmosphere Thomas Gautier, Nathalie Carrasco, Arnaud Buch, Cyril Szopa, Ella Sciamma-O’Brien, Guy Cernogora
Nitrile gas chemistry in Titan atmosphere Thomas Gautier, Nathalie Carrasco, Arnaud Buch, Cyril Szopa, Ella Sciamma-O’Brien, Guy Cernogora To cite this version: Thomas Gautier, Nathalie Carrasco, Arnaud Buch, Cyril Szopa, Ella Sciamma-O’Brien, et al.. Nitrile gas chemistry in Titan atmosphere. Icarus, Elsevier, 2011, 213 (2), pp.625-635. 10.1016/j.icarus.2011.04.005. hal-00756910 HAL Id: hal-00756910 https://hal.archives-ouvertes.fr/hal-00756910 Submitted on 24 Nov 2012 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Accepted Manuscript Nitrile gas chemistry in Titan atmosphere Thomas Gautier, Nathalie Carrasco, Arnaud Buch, Cyril Szopa, Ella Sciamma- O’Brien, Guy Cernogora PII: S0019-1035(11)00126-6 DOI: 10.1016/j.icarus.2011.04.005 Reference: YICAR 9778 To appear in: Icarus Received Date: 6 January 2011 Revised Date: 7 April 2011 Accepted Date: 7 April 2011 Please cite this article as: Gautier, T., Carrasco, N., Buch, A., Szopa, C., Sciamma-O’Brien, E., Cernogora, G., Nitrile gas chemistry in Titan atmosphere, Icarus (2011), doi: 10.1016/j.icarus.2011.04.005 This is a PDF file of an unedited manuscript that has been accepted for publication. -

Hazardous Chemicals Handbook
Hazardous Chemicals Handbook Hazardous Chemicals Handbook Second edition Phillip Carson PhD MSc AMCT CChem FRSC FIOSH Head of Science Support Services, Unilever Research Laboratory, Port Sunlight, UK Clive Mumford BSc PhD DSc CEng MIChemE Consultant Chemical Engineer Oxford Amsterdam Boston London New York Paris San Diego San Francisco Singapore Sydney Tokyo Butterworth-Heinemann An imprint of Elsevier Science Linacre House, Jordan Hill, Oxford OX2 8DP 225 Wildwood Avenue, Woburn, MA 01801-2041 First published 1994 Second edition 2002 Copyright © 1994, 2002, Phillip Carson, Clive Mumford. All rights reserved The right of Phillip Carson and Clive Mumford to be identified as the authors of this work has been asserted in accordance with the Copyright, Designs and Patents Act 1988 No part of this publication may be reproduced in any material form (including photocopying or storing in any medium by electronic means and whether or not transiently or incidentally to some other use of this publication) without the written permission of the copyright holder except in accordance with the provisions of the Copyright, Designs and Patents Act 1988 or under the terms of a licence issued by the Copyright Licensing Agency Ltd, 90 Tottenham Court Road, London, England W1T 4LP. Applications for the copyright holder’s written permission to reproduce any part of this publication should be addressed to the publishers British Library Cataloguing in Publication Data A catalogue record for this book is available from the British Library Library of Congress Cataloguing