Infrared Spectroscopy of Dilute HOD In
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Using Visible and Near-Infrared Reflectance Spectroscopy and Differential Scanning Calorimetry to Study Starch, Protein, and Temperature Effects on Bread Staling
Using Visible and Near-Infrared Reflectance Spectroscopy and Differential Scanning Calorimetry to Study Starch, Protein, and Temperature Effects on Bread Staling Feng Xie,1 Floyd E. Dowell,2,3 and Xiuzhi S. Sun1 ABSTRACT Cereal Chem. 81(2):249–254 Starch, protein, and temperature effects on bread staling were inves- retrograded amylose-lipid complex was limited. Two important wave- tigated using visible and near-infrared spectroscopy (NIRS) and differ- lengths, 550 nm and 1,465 nm, were key for NIRS to successfully classify ential scanning calorimetry (DSC). Bread staling was mainly due to the starch-starch (SS) and starch-protein (SP) bread based on different amylopectin retrogradation. NIRS measured amylopectin retrogradation colors and protein contents in SS and SP. Low temperature dramatically accurately in different batches. Three important wavelengths, 970 nm, accelerated the amylopectin retrogradation process. Protein retarded bread 1,155 nm, and 1,395 nm, were associated with amylopectin retrogra- staling, but not as much as temperature. The starch and protein interaction dation. NIRS followed moisture and starch structure changes when was less important than the starch retrogradation. Protein hindered the amylopectin retrograded. The amylose-lipid complex changed little from bread staling process mainly by diluting starch and retarding starch retro- one day after baking. The capability of NIRS to measure changes in the gradation. Bread staling is a complex process that occurs during bread and starch (Osborne and Douglas 1981; Davies and Miller 1988; storage. It is a progressive deterioration of qualities such as taste, Millar et al 1996). Therefore, NIRS has the potential to provide fun- firmness, etc. -

Basic Keyword List
NOTICE TO AUTHORS 2008 Basic Keyword list A Antibodies Bismuth Chalcogens Ab initio calculations Antifungal agents Block copolymers Chaperone proteins Absorption Antigens Bond energy Charge carrier injection Acidity Antimony Bond theory Charge transfer Actinides Antisense agents Boranes Chelates Acylation Antitumor agents Borates Chemical vapor deposition Addition to alkenes Antiviral agents Boron Chemical vapor transport Addition to carbonyl com- Aqueous-phase catalysis Bridging ligands Chemisorption pounds Arene ligands Bromine Chemoenzymatic synthesis Adsorption Arenes Brønsted acids Chemoselectivity Aerobic oxidation Argon Chiral auxiliaries Aggregation Aromatic substitution C Chiral pool Agostic interactions Aromaticity C-C activation Chiral resolution Alanes Arsenic C-C bond formation Chirality Alcohols Arylation C-C coupling Chlorine Aldehydes Aryl halides C-Cl bond activation Chromates Aldol reaction Arynes C-Glycosides Chromium Alkali metals As ligands C-H activation Chromophores Alkaline earth metals Asymmetric amplification C1 building blocks Circular dichroism Alkaloids Asymmetric catalysis Cadmium Clathrates Alkanes Asymmetric synthesis Cage compounds Clays Alkene ligands Atmospheric chemistry Calcium Cleavage reactions Alkenes Atom economy Calixarenes Cluster compounds Alkylation Atropisomerism Calorimetry Cobalamines Alkyne ligands Aurophilicity Carbanions Cobalt Alkynes Autocatalysis Carbene homologues Cofactors Alkynylation Automerization Carbene ligands Colloids Allenes Autoxidation Carbenes Combinatorial chemistry Allosterism -

Photochemistry of Highly Excited States R
COMMENTARY COMMENTARY Photochemistry of highly excited states R. D. Levinea,b,c,1 These days on earth we are photochemically reasonably A forceful illustration of the benefits of the synergy of well shielded. The far UV radiation is filtered by the ozone experiment and theory in understanding the detailed and other components of the atmosphere. In the iono- electronic and nuclear dynamics in highly excited states is sphere and beyond there is much challenging chemistry provided by the very recent work reported by Peters et al. induced by such higher-energy photons. The shielding by (8). Methyl azide is pumped by an 8-eV vacuum UV (VUV) the atmosphere also makes demands on such experi- pulse of 10-fs duration and probed by a second 1.6-eV in- ments in the laboratory. There is, however, recent interest frared (IR) pulse that ionizes the molecule. The experiment largely driven by technology that makes higher resolution as seen through the eyes of a theorist is shown in Fig. 1. The possible. The experimental technology is advanced light pump pulse prepares a single excited electronic state, la- sources that offer far higher intensities, sharper wave- beled as S8, that theory shows has a mixed valence and length definition or, complementarily, short-duration Rydberg character. It is unusual that a single excited state pulses. The two alternatives make a whole because of is accessed at such a high energy. The high-level computa- the quantum mechanical time–energy uncertainty princi- tions reported by Peters et al. are quite clear that the ple that makes short pulses necessarily broad in energy. -

Comparison Between FTIR and Boehm Titration for Activated Carbon Functional Group Quantification
Comparison Between FTIR and Boehm Titration for Activated Carbon Functional Group Quantification Chad Spreadbury, Regina Rodriguez, and David Mazyck College of Engineering, University of Florida Activated carbon (AC) is a proven effective adsorbent of contaminants in the air and water phases. This effectiveness is due to the large surface area of AC which hosts functional groups. Particularly, oxygen functional groups (e.g. carboxyls, lactones, phenols, carbonyls) have been noted as important in mercury removal. Hence, determining the quantities of these groups is crucial when AC is used for this purpose. Currently, Boehm titration is the most common method for determining the number of these functional groups. However, this test method is susceptible to high user error and require a lengthy procedure and run time. On the other hand, Fourier transform infrared (FTIR) spectroscopy excels at analyzing the functionality of an AC surface. This study investigates whether or not a correlation between Boehm titration and FTIR can be determined using quantitative and qualitative assessment. The results indicate that while using the current methodology for Boehm titrations and FTIR analysis, there is no clear correlation. However, this study does not rule out that a correlation may indeed exist between these test methods that can be elucidated with improved methodology that accounts for the unique physical and chemical characteristics of carbon. INTRODUCTION mercury (Hg0) to create oxidized mercury (Hg2+), which then undergoes chemisorption with the AC surface. ctivated carbon is used globally as an effective Boehm titrations and Fourier transform infrared (FTIR) adsorbent for removing contaminants like mercury spectroscopy are two common testing procedures for A(Hg) from air and water phases. -

Laboratory Astrophysics: from Observations to Interpretation
April 14th – 19th 2019 Jesus College Cambridge UK IAU Symposium 350 Laboratory Astrophysics: From Observations to Interpretation Poster design by: D. Benoit, A. Dawes, E. Sciamma-O’Brien & H. Fraser Scientific Organizing Committee: Local Organizing Committee: Farid Salama (Chair) ★ P. Barklem ★ H. Fraser ★ T. Henning H. Fraser (Chair) ★ D. Benoit ★ R Coster ★ A. Dawes ★ S. Gärtner ★ C. Joblin ★ S. Kwok ★ H. Linnartz ★ L. Mashonkina ★ T. Millar ★ D. Heard ★ S. Ioppolo ★ N. Mason ★ A. Meijer★ P. Rimmer ★ ★ O. Shalabiea★ G. Vidali ★ F. Wa n g ★ G. Del-Zanna E. Sciamma-O’Brien ★ F. Salama ★ C. Wa lsh ★ G. Del-Zanna For more information and to contact us: www.astrochemistry.org.uk/IAU_S350 [email protected] @iaus350labastro 2 Abstract Book Scheduley Sunday 14th April . Pg. 2 Monday 15th April . Pg. 3 Tuesday 16th April . Pg. 4 Wednesday 17th April . Pg. 5 Thursday 18th April . Pg. 6 Friday 19th April . Pg. 7 List of Posters . .Pg. 8 Abstracts of Talks . .Pg. 12 Abstracts of Posters . Pg. 83 yPlenary talks (40') are indicated with `P', review talks (30') with `R', and invited talks (15') with `I'. Schedule Sunday 14th April 14:00 - 17:00 REGISTRATION 18:00 - 19:00 WELCOME RECEPTION 19:30 DINNER BAR OPEN UNTIL 23:00 Back to Table of Contents 2 Monday 15th April 09:00 { 10:00 REGISTRATION 09:00 WELCOME by F. Salama (Chair of SOC) SESSION 1 CHAIR: F. Salama 09:15 E. van Dishoeck (P) Laboratory astrophysics: key to understanding the Universe From Diffuse Clouds to Protostars: Outstanding Questions about the Evolution of 10:00 A. -

Application of High Performance Liquid Chromatography (HPLC) and Fourier Transform Infrared (FTIR) Spectroscopy to Swiss-Typ
Application of High Performance Liquid Chromatography (HPLC) and Fourier Transform Infrared (FTIR) Spectroscopy to Swiss-type Cheese Split Defects THESIS Presented in Partial Fulfillment of the Requirements for the Master of Science Degree in the Graduate School of The Ohio State University By Feifei Hu Graduate Program in Food Science and Technology The Ohio State University 2012 Master's Examination Committee: Dr. W. James Harper, Advisor Dr. Luis E. Rodriguez-Saona Dr. Michael E. Mangino Copyright Feifei Hu 2012 Abstract Splits, slits and cracks are a continuing problem in the US Swiss Cheese industry and cause downgrading of the cheese, bringing economic loss. The defects generally occur during secondary fermentation in cold storage, which is known to be an issue caused by many factors. Many chemical and physical changes can be indicators to help predicting the defects. Various techniques have been applied to test different compounds in cheese. HPLC is one of the reference methods which have been applied in cheese chemistry for the past decades. Its application involves in the qualification and quantification of organic acids, amino acids and peptides, and sugars in dairy products. However, limited research has been conducted on a rapid and easy testing regimen to investigate split defects in Swiss-type cheese. For over fifty years, researchers have investigated the application of Fourier Transform Infrared (FTIR) Spectroscopy in various food samples, including cheeses and other dairy products because FTIR spectroscopy gives fast, simple and comprehensive detection of chemical compounds. There is great potential for applying FTIR spectroscopy in developing a rapid test method to detect and predict the occurrence of split defects. -
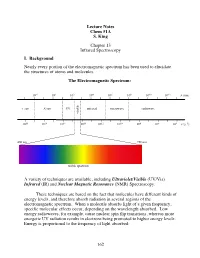
162 Lecture Notes Chem 51A S. King Chapter 13 Infrared Spectroscopy I
Lecture Notes Chem 51A S. King Chapter 13 Infrared Spectroscopy I. Background Nearly every portion of the electromagnetic spectrum has been used to elucidate the structures of atoms and molecules. The Electromagnetic Spectrum: 10!2 100 102 104 106 108 1010 1012 $ (nm) "!ray X-ray UV infrared microwave radiowave visible 1020 1018 1016 1014 1012 1010 108 106 104 # (s!1) 400 nm 750 nm ! visible spectrum A variety of techniques are available, including Ultraviolet/Visible (UV/Vis) Infrared (IR) and Nuclear Magnetic Resonance (NMR) Spectroscopy. These techniques are based on the fact that molecules have different kinds of energy levels, and therefore absorb radiation in several regions of the electromagnetic spectrum. When a molecule absorbs light of a given frequency, specific molecular effects occur, depending on the wavelength absorbed. Low energy radiowaves, for example, cause nuclear spin flip transitions, whereas more energetic UV radiation results in electrons being promoted to higher energy levels. Energy is proportional to the frequency of light absorbed: 162 Molecular effects associated with different regions of the EM spectrum: Wavelength (!) Energy/mole Molecular effects 10"10 meter gamma rays 106 kcal 10"8 meter X-rays 104 kcal ionization vaccum UV 102 kcal near UV electronic transitions 10"6 meter visible 10 kcal infrared 1 kcal molecular vibrations 10"4 meter (IR) 10"2 kcal 10"2 meter microwave rotational motion 10"4 kcal 0 10 meter radio 10"6 kcal nuclear spin transitions 2 10 meter II. IR Spectroscopy IR radiation causes groups of atoms to vibrate with respect to the bonds that connect them. -
Infrared and UV-Visible Time-Resolved Techniques for the Study of Tetrapyrrole-Based Proteins
Infrared and UV-visible time-resolved techniques for the study of tetrapyrrole-based proteins A thesis submitted to The University of Manchester for the degree of Doctor of Philosophy (PhD) in the Faculty of Life Sciences 2013 Henry John Russell Contents List of Tables .......................................................................................................................................................................... 5 List of Figures ........................................................................................................................................................................ 6 Abstract ...................................................................................................................................................................................10 Declaration ............................................................................................................................................................................11 Copyright Statement ........................................................................................................................................................11 Acknowledgements ..........................................................................................................................................................12 Abbreviations and Symbols ..........................................................................................................................................13 Preface and Thesis Structure .......................................................................................................................................16 -

And Raman Spectroscopy IR and Raman Spectroscopy Measure the Energy Difference of Vibrational Energy Levels in Molecules, They Are Energy Sensitive Methods
Infrared (IR) and Raman Spectroscopy IR and Raman spectroscopy measure the energy difference of vibrational energy levels in molecules, they are energy sensitive methods. They are based on interactions of electromagnetic radiation and material but the main differences between these two spectroscopic techniques are the physical effects. For infrared spectroscopy the absorption process of polychromatic IR radiation due to the molecule cause a change in the dipole moment of the molecule and the molecule will be translated from a ground state to a vibrational excited state. Whereas the monochromatic radiation, for Raman spectroscopy, is inelastic scattered at the electron shell of the molecule which changes the molecular polarizability and the radiation will be shifted in frequency (light source: UV, VIS or NIR lasers). The “Raman effect” was discovered by the Indian physicist, C. V. Raman inn 1928. Selection rules: Whether or not a vibrational mode is active or inactive for one of these complementary (inversely related) techniques depends on the symmetry of the molecules. For example, homonuclear diatomic molecules are not IR active, because they have no dipole moment, but they are Raman active. Because of the stretching and contraction of the bond changes the interactions between the electrons and nuclei, this causes a change of polarizability. For highly symmetric polyatomic molecules possessing a center of inversion, the bands are IR active (Raman inactive) for asymmetric vibrations to i and for symmetric vibrations to i the bands are Raman active (IR inactive). A mode can be IR active, Raman inactive and vice-versa however not at the same time. This fact is named as mutual exclusion rule. -

Angwcheminted, 2000, 39, 2587-2631.Pdf
REVIEWS Femtochemistry: Atomic-Scale Dynamics of the Chemical Bond Using Ultrafast Lasers (Nobel Lecture)** Ahmed H. Zewail* Over many millennia, humankind has biological changes. For molecular dy- condensed phases, as well as in bio- thought to explore phenomena on an namics, achieving this atomic-scale res- logical systems such as proteins and ever shorter time scale. In this race olution using ultrafast lasers as strobes DNA structures. It also offers new against time, femtosecond resolution is a triumph, just as X-ray and electron possibilities for the control of reactivity (1fs 10À15 s) is the ultimate achieve- diffraction, and, more recently, STM and for structural femtochemistry and ment for studies of the fundamental and NMR spectroscopy, provided that femtobiology. This anthology gives an dynamics of the chemical bond. Ob- resolution for static molecular struc- overview of the development of the servation of the very act that brings tures. On the femtosecond time scale, field from a personal perspective, en- about chemistryÐthe making and matter wave packets (particle-type) compassing our research at Caltech breaking of bonds on their actual time can be created and their coherent and focusing on the evolution of tech- and length scalesÐis the wellspring of evolution as a single-molecule trajec- niques, concepts, and new discoveries. the field of femtochemistry, which is tory can be observed. The field began the study of molecular motions in the with simple systems of a few atoms and Keywords: femtobiology ´ femto- hitherto unobserved ephemeral transi- has reached the realm of the very chemistry ´ Nobel lecture ´ physical tion states of physical, chemical, and complex in isolated, mesoscopic, and chemistry ´ transition states 1. -

Femtochemistry' Ahmed Zewail Wins the 1999 Nobel Prize in Chemistry
GENERAL I ARTICLE The Man Behind 'Femtochemistry' Ahmed Zewail Wins the 1999 Nobel Prize in Chemistry Puspendu Kumar Das He hails from the land of the Pharaohs; he came to the United States, like many, to seek higher education and today he is at the top of his achievements. He is Ahmed H Zewail, Linus Pauling Chair of Chemical Physics at the California Institute of Tech nology. He has just been honoured with the Nobel Prize in Chemistry by the Royal Swedish Academy of Sciences on Octo ber 12, 1999, 'for his studies of the transition state of chemical Puspendu Kumar Das reactions using femtosecond spectroscopy'. Bond formation and studies and teaches bond breaking are the two most fundamental concepts that physical chemistry at the department of inorganic make chemistry. Zewail studied these two processes using ultra and physical chemistry short techniques on the time scale on which they actually occur. in the Indian Institute of Science, Bangalore. His The Man current research interests include gas Born February 26, 1946 in Egypt, Ahmed H Zewail (Figure 1) phase chemical kinetics, received his BS and MS degrees from Alexandria University second order nonlinear optics, and lipid-protein which was, perhaps, once the greatest place for learning on interaction. earth. He then moved to the University of Pennsylvania, Phila delphia in USA and obtained a PhD degree in 1974 working with R M Hochstrasser. Following his postdoctoral work with Charles Harris at the University of California at Berkeley, he joined the chemistry department of Caltech in 1976. Figure 1. A stamp size He was tenured in two years and became a full portrait (truly) of professor in 1982. -

Fourier Transform Infrared Spectroscopy
FOURIER TRANSFORM INFRARED SPECTROSCOPY Purpose and Scope: This document describes the procedures and policies for using the MSE Bruker FTIR Spectrometer. The scope of this document is to establish user procedures. Instrument maintenance and repair are outside the scope of this document. Responsibilities: This document is maintained by the department Lab manager. The Lab Manager is responsible for general maintenance and for arranging repair when necessary. If you feel that the instrument is in need of repair or is not operating correctly, please notify the Lab Manager immediately. The Lab Manger will operate the instruments according to the procedures set down in this document and will provide instruction and training to users within the department. Users are responsible for using the instrument described according to these procedures. These procedures assume that the user has had at least one training session. Warnings and precautions: • When using the EGA-TGA Cell remember that it is hot. The cell and the line from the TGA to the cell are operated at 200C. • When using the EGA-TGA cell please remember to turn off the heater by pressing the down arrow on the controller until the set point reads “off”. You will be shown how to do this during training. • User are NOT cleared to change out fixtures. Only lab personnel are allowed to install or remove the ATR, the pellet holder, the EGA-TGA Cell or any other fixtures. We recommend that you notify lab personnel the day before your scheduled measurement to ensure it is ready. • For measuring KBr pellets: Users are expected to make/have their own pellet equipment.