WHO Guidelines for the Production, Control and Regulation of Snake Antivenom Immunoglobulins
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
Snake Bite Protocol
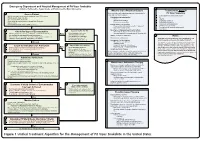
Lavonas et al. BMC Emergency Medicine 2011, 11:2 Page 4 of 15 http://www.biomedcentral.com/1471-227X/11/2 and other Rocky Mountain Poison and Drug Center treatment of patients bitten by coral snakes (family Ela- staff. The antivenom manufacturer provided funding pidae), nor by snakes that are not indigenous to the US. support. Sponsor representatives were not present dur- At the time this algorithm was developed, the only ing the webinar or panel discussions. Sponsor represen- antivenom commercially available for the treatment of tatives reviewed the final manuscript before publication pit viper envenomation in the US is Crotalidae Polyva- ® for the sole purpose of identifying proprietary informa- lent Immune Fab (ovine) (CroFab , Protherics, Nash- tion. No modifications of the manuscript were requested ville, TN). All treatment recommendations and dosing by the manufacturer. apply to this antivenom. This algorithm does not con- sider treatment with whole IgG antivenom (Antivenin Results (Crotalidae) Polyvalent, equine origin (Wyeth-Ayerst, Final unified treatment algorithm Marietta, Pennsylvania, USA)), because production of The unified treatment algorithm is shown in Figure 1. that antivenom has been discontinued and all extant The final version was endorsed unanimously. Specific lots have expired. This antivenom also does not consider considerations endorsed by the panelists are as follows: treatment with other antivenom products under devel- opment. Because the panel members are all hospital- Role of the unified treatment algorithm -

Freshwater Fishes
WESTERN CAPE PROVINCE state oF BIODIVERSITY 2007 TABLE OF CONTENTS Chapter 1 Introduction 2 Chapter 2 Methods 17 Chapter 3 Freshwater fishes 18 Chapter 4 Amphibians 36 Chapter 5 Reptiles 55 Chapter 6 Mammals 75 Chapter 7 Avifauna 89 Chapter 8 Flora & Vegetation 112 Chapter 9 Land and Protected Areas 139 Chapter 10 Status of River Health 159 Cover page photographs by Andrew Turner (CapeNature), Roger Bills (SAIAB) & Wicus Leeuwner. ISBN 978-0-620-39289-1 SCIENTIFIC SERVICES 2 Western Cape Province State of Biodiversity 2007 CHAPTER 1 INTRODUCTION Andrew Turner [email protected] 1 “We live at a historic moment, a time in which the world’s biological diversity is being rapidly destroyed. The present geological period has more species than any other, yet the current rate of extinction of species is greater now than at any time in the past. Ecosystems and communities are being degraded and destroyed, and species are being driven to extinction. The species that persist are losing genetic variation as the number of individuals in populations shrinks, unique populations and subspecies are destroyed, and remaining populations become increasingly isolated from one another. The cause of this loss of biological diversity at all levels is the range of human activity that alters and destroys natural habitats to suit human needs.” (Primack, 2002). CapeNature launched its State of Biodiversity Programme (SoBP) to assess and monitor the state of biodiversity in the Western Cape in 1999. This programme delivered its first report in 2002 and these reports are updated every five years. The current report (2007) reports on the changes to the state of vertebrate biodiversity and land under conservation usage. -

Molecular Characterization of Three Novel Phospholipase A2 Proteins from the Venom of Atheris Chlorechis, Atheris Nitschei and Atheris Squamigera
toxins Article Molecular Characterization of Three Novel Phospholipase A2 Proteins from the Venom of Atheris chlorechis, Atheris nitschei and Atheris squamigera He Wang 1,*, Xiaole Chen 2,*, Mei Zhou 3, Lei Wang 3, Tianbao Chen 3 and Chris Shaw 3 1 School of Integrative Medicine, Fujian University of Traditional Chinese Medicine, No.1 Qiu Yang Road, Shangjie Town, Fuzhou 350122, Fujian, China 2 School of Pharmacy, Fujian Medical University, No.1 Xueyuan Road, Shangjie Town, Fuzhou 350004, Fujian, China 3 Natural Drug Discovery Group, School of Pharmacy, Queen’s University Belfast, University Road, Belfast BT7 1NN, UK; [email protected] (M.Z.); [email protected] (L.W.); [email protected] (T.C.); [email protected] (C.S.) * Correspondence: [email protected] or [email protected] (H.W.); [email protected] (X.C.); Tel.: +86-591-2286-1151 (H.W.); +86-591-2286-2692 (X.C.) Academic Editor: Bryan Grieg Fry Received: 24 February 2016; Accepted: 20 May 2016; Published: 1 June 2016 Abstract: Secretory phospholipase A2 (sPLA2) is known as a major component of snake venoms and displays higher-order catalytic hydrolysis functions as well as a wide range of pathological effects. Atheris is not a notoriously dangerous genus of snakes although there are some reports of fatal cases after envenomation due to the effects of coagulation disturbances and hemorrhaging. Molecular characterization of Atheris venom enzymes is incomplete and there are only a few reports in the literature. Here, we report, for the first time, the cloning and characterization of three novel cDNAs encoding phospholipase A2 precursors (one each) from the venoms of the Western bush viper (Atheris chlorechis), the Great Lakes bush viper (Atheris nitschei) and the Variable bush viper (Atheris squamigera), using a “shotgun cloning” strategy. -

Historia Natural De Golfito VERDE NATIONAL PARK • José Sancho En El Inbioparque • Árboles De Costa Rica, Vol
Historia natural de Golfi to Costa Rica Jorge Lobo Federico Bolaños Editores científicos 508.728 H673b Historia natural de Golfito – Costa Rica / Editado por Jorge Lobo Segura y Federico Bolaños Vives.- Santo Domingo de Heredia, Costa Rica: Instituto Nacional de Biodiversidad, INBio, 2005 264 p.; ils.; 15x 22,8 cm Incluye fotografías a color ISBN 9968-927-07-4 1. Ciencias naturales. 2. Conservación de los recursos naturales. 3. Biodiversidad (Golfito – Costa Rica). I. Lobo Segura, Jorge, Ed. científico. II. Bolaños Vives, Federico, Ed. científico. III Título Esta publicación se hizo gracias a la colaboración financiera del proyecto Desarrollo del conocimiento y uso sostenible de la biodiversidad en Costa Rica, del Gobierno de los Países Bajos. Gerente editorial: Fabio Rojas Carballo Editora: Diana Ávila Solera Diseño y diagramación: Esteban Ocampo Cubero Revisión científica: Jorge Lobo, Federico Bolaños Primera edición 2005 © Instituto Nacional de Biodiversidad (INBio) Hecho el depósito de ley. Reservados todos los derechos. Prohibida la reproducción total o parcial de este libro. Hecho en Costa Rica por la Editorial INBio Contenido Agradecimientos . 7 Prefacio . 9 Lista de autores participantes . 11 Introducción . 13 Características geográficas de la región de Golfito . 19 Historia de la región de Golfo Dulce . 25 Ecosistemas acuáticos Plancton . 45 Manglares . 55 Los ríos de la cuenca del Golfo Dulce . 67 Diversidad, ecología e importancia de los insectos acuáticos . 81 Plantas Aspectos generales del bosque del Refugio Nacional de Vida Silvestre Golfito . 97 La estructura vertical del bosque de Golfito . 107 Las platanillas del Refugio Nacional de Vida Silvestre Golfito . 119 Insectos Los machos de la libélula Hetaerina en Golfito: ¿por qué pelean tanto? . -

Historia Natural Y Cultural De La Región Del Golfo Dulce, Costa Rica
Natural and Cultural History of the Golfo Dulce Region, Costa Rica Historia natural y cultural de la región del Golfo Dulce, Costa Rica Anton WEISSENHOFER , Werner HUBER , Veronika MAYER , Susanne PAMPERL , Anton WEBER , Gerhard AUBRECHT (scientific editors) Impressum Katalog / Publication: Stapfia 88 , Zugleich Kataloge der Oberösterreichischen Landesmuseen N.S. 80 ISSN: 0252-192X ISBN: 978-3-85474-195-4 Erscheinungsdatum / Date of deliVerY: 9. Oktober 2008 Medieninhaber und Herausgeber / CopYright: Land Oberösterreich, Oberösterreichische Landesmuseen, Museumstr.14, A-4020 LinZ Direktion: Mag. Dr. Peter Assmann Leitung BiologieZentrum: Dr. Gerhard Aubrecht Url: http://WWW.biologieZentrum.at E-Mail: [email protected] In Kooperation mit dem Verein Zur Förderung der Tropenstation La Gamba (WWW.lagamba.at). Wissenschaftliche Redaktion / Scientific editors: Anton Weissenhofer, Werner Huber, Veronika MaYer, Susanne Pamperl, Anton Weber, Gerhard Aubrecht Redaktionsassistent / Assistant editor: FritZ Gusenleitner LaYout, Druckorganisation / LaYout, printing organisation: EVa Rührnößl Druck / Printing: Plöchl-Druck, Werndlstraße 2, 4240 Freistadt, Austria Bestellung / Ordering: http://WWW.biologieZentrum.at/biophp/de/stapfia.php oder / or [email protected] Das Werk einschließlich aller seiner Teile ist urheberrechtlich geschütZt. Jede VerWertung außerhalb der en - gen GrenZen des UrheberrechtsgesetZes ist ohne Zustimmung des Medieninhabers unZulässig und strafbar. Das gilt insbesondere für VerVielfältigungen, ÜbersetZungen, MikroVerfilmungen soWie die Einspeicherung und Verarbeitung in elektronischen SYstemen. Für den Inhalt der Abhandlungen sind die Verfasser Verant - Wortlich. Schriftentausch erWünscht! All rights reserVed. No part of this publication maY be reproduced or transmitted in anY form or bY anY me - ans Without prior permission from the publisher. We are interested in an eXchange of publications. Umschlagfoto / CoVer: Blattschneiderameisen. Photo: AleXander Schneider. -

Cassini Observations of Saturn's Irregular Moons
EPSC Abstracts Vol. 12, EPSC2018-103-1, 2018 European Planetary Science Congress 2018 EEuropeaPn PlanetarSy Science CCongress c Author(s) 2018 Cassini Observations of Saturn's Irregular Moons Tilmann Denk (1) and Stefano Mottola (2) (1) Freie Universität Berlin, Germany ([email protected]), (2) DLR Berlin, Germany 1. Introduction two prograde irregulars are slower than ~13 h, while the periods of all but two retrogrades are faster than With the ISS-NAC camera of the Cassini spacecraft, ~13 h. The fastest period (Hati) is much slower than we obtained photometric lightcurves of 25 irregular the disruption rotation barrier for asteroids (~2.3 h), moons of Saturn. The goal was to derive basic phys- indicating that Saturn's irregulars may be rubble piles ical properties of these objects (like rotational periods, of rather low densities, possibly as low as of comets. shapes, pole-axis orientations, possible global color variations, ...) and to get hints on their formation and Table: Rotational periods of 25 Saturnian irregulars evolution. Our campaign marks the first utilization of an interplanetary probe for a systematic photometric Moon Approx. size Rotational period survey of irregular moons. name [km] [h] Hati 5 5.45 ± 0.04 The irregular moons are a class of objects that is very Mundilfari 7 6.74 ± 0.08 distinct from the inner moons of Saturn. Not only are Loge 5 6.9 ± 0.1 ? they more numerous (38 versus 24), but also occupy Skoll 5 7.26 ± 0.09 (?) a much larger volume within the Hill sphere of Suttungr 7 7.67 ± 0.02 Saturn. -

Bulletin 107 FRONT COVER FIXED.Indd
The HERPETOLOGICAL BULLETIN Number 107 – Spring 2009 PUBLISHED BY THE BRITISH HERPETOLOGICAL SOCIETY THE HERPETOLOGICAL BULLETIN Contents NEWS REPO R TS . 1 RESEA R CH AR TICLES Possible decline in an American Crocodile (Crocodylus acutus) population on Turneffe Atoll, Belize Thomas R. Rainwater and Steven G. Platt ............................. 3 Range extension of Kaestlea beddomeii (Boulenger, 1887) (in part) (Reptilia: Sauria: Scincidae) S. R. Ganesh and P. Gowri Shankar ................................. 12 The herpetofauna of Koanaka South and adjacent regions, Ngamiland, Botswana Aaron M. Bauer, Alicia M. Kennedy, Patrick J. Lewis, Monte L. Thies and . Mohutsiwa Gabadirwe . 16 Prodichotomy in the snake Oreocryptophis porhyraceus coxi (Schulz & Helfenberger, 1998) (Serpentes: Colubridae) David Jandzik . 27 Reptiles and amphibians from the Kenyan coastal hinterland N. Thomas Håkansson . 30 NATU R AL HISTO R Y NOTES Nucras taeniolata Smith, 1838 (Striped Sandveld Lizard) (Sauria, Lacertidae): additional records William R. Branch and M. Burger . 40 Oreocryptophis porphyraceus coxi (Thai Bamboo Ratsnake): pattern abnormality David Jandzik . 41 Norops sagrei (Brown Anole): pathology and endoparasite Gerrut Norval, Charles R. Bursey, Stephen R. Goldberg, Chun-Liang Tung and Jean-Jay Mao . 42 - Registered Charity No. 205666 - - Registered Charity No. 205666 - THE HERPETOLOGICAL BULLETIN The Herpetological Bulletin is produced quarterly and publishes, in English, a range of articles concerned with herpetology. These include society news, selected news reports, full-length papers of a semi- technical nature, new methodologies, natural history notes, book reviews, letters from readers and other items of general herpetological interest. Emphasis is placed on natural history, conservation, captive breeding and husbandry, veterinary and behavioural aspects. Articles reporting the results of experimental research, descriptions of new taxa, or taxonomic revisions should be submitted to The Herpetological Journal (see inside back cover for Editor’s address). -

Annual Report 2017
3 CONTACT DETAILS Dean Prof Danie Vermeulen +27 51 401 2322 [email protected] MARKETING MANAGER ISSUED BY Ms Elfrieda Lötter Faculty of Natural and Agricultural Sciences +27 51 401 2531 University of the Free State [email protected] EDITORIAL COMPILATION PHYSICAL ADDRESS Ms Elfrieda Lötter Room 9A, Biology Building, Main Campus, Bloemfontein LANGUAGE REVISION Dr Cindé Greyling and Elize Gouws POSTAL ADDRESS University of the Free State REVISION OF BIBLIOGRAPHICAL DATA PO Box 339 Dr Cindé Greyling Bloemfontein DESIGN, LAYOUT South Africa )LUHÀ\3XEOLFDWLRQV 3W\ /WG 9300 PRINTING Email: [email protected] SA Printgroup )DFXOW\ZHEVLWHZZZXIVDF]DQDWDJUL 4 NATURAL AND AGRICULTURAL SCIENCES REPORT 2017 CONTENT PREFACE Message from the Dean 7 AGRICULTURAL SCIENCES Agricultural Economics 12 Animal, Wildlife and Grassland Sciences 18 Plant Sciences 26 Soil, Crop and Climate Sciences 42 BUILDING SCIENCES Architecture 50 Quantity Surveying and Construction Management 56 8UEDQDQG5HJLRQDO3ODQQLQJ NATURAL SCIENCES Chemistry 66 Computer Sciences and Informatics 80 Consumer Sciences 88 Genetics 92 Geography 100 Geology 106 Mathematical Statistics and Actuarial Science 112 Mathematics and Applied Mathematics 116 Mathematics 120 0LFURELDO%LRFKHPLFDODQG)RRG%LRWHFKQRORJ\ Physics 136 Zoology and Entomology 154 5 Academic Centres Disaster Management Training and Education Centre of Africa - DiMTEC 164 Centre for Environmental Management - CEM 170 Centre for Microscopy 180 6XVWDLQDEOH$JULFXOWXUH5XUDO'HYHORSPHQWDQG([WHQVLRQ Paradys Experimental Farm 188 Engineering Sciences 192 Institute for Groundwater Studies 194 ACADEMIC SUPPORT UNITS Electronics Division 202 Instrumentation 206 STATISTICAL DATA Statistics 208 LIST OF ACRONYMS List of Acronyms 209 6 NATURAL AND AGRICULTURAL SCIENCES REPORT 2017 0(66$*( from the '($1 ANNUAL REPORT 2016 will be remembered as one of the worst ±ZKHUHHDFKELQFRXOGFRQWDLQDXQLTXHSURGXFWDQG years for tertiary education in South Africa due once a product is there, it remains. -

Sudan Snakes
NOTES ON SUDAN SNAKES A GUIDE TO THE SPECIES REPRESENTED IN THE COLLECTION IN THE NATURAL HISTORY MUSEUM KHARTOUM BY N. L. CORKILL, M.D. Sudan Medical Service. SUDAN GOVERNMENT MUSEUM (NATURAL HISTORY) PUBLICATION No. 3 AUGUST, 1935 PRICE - - - P:T. 10(2/-). NOTES ON SUDAN SNAKES A GUIDE TO THE SPECIES REPRESENTED IN THE COLLECTION IN THE NATURAL HISTORY MUSEUM KHARTOUM BY N. L. GORKILL, M.D. Sudan Medical Service. SUDAN GOVERNMENT MUSEUM (NATURAL HISTORY) PUBLICATION No. 3 AUGUST, 1935 PRICE - - - P.T. 10(2/-). PRINTED BY M*CORQUODALE & CO. J CONTENTS. PAGE Foreword ... ... ... ... ... ... ••• ••• 4 Preface ... 5 Introduction... ... ... ... ... ... ... ... 6 Systematic Index ... ... ... ... ... ... ... 9 Family Typhlopidge, the Blind Snakes 11 Family Leptotyphlopidse, the Earth Snakes ... ... ... n Family Boidae, the Boas and Pythons ... ... ... ... 12 Family Colubridae, the Colubrids 14 (a) Series Aglypha, the Fangless Colubrids ... ... 14 (b) Series Opisthoglypha, the Back-fanged Colubrids ... 19 Family Elapidae, the Cobras 23 Family Viperidas, the Vipers ... ... ... ... ... 26 Key to the Identification of Sudanese Thanat ophidians ... 31 Snake Bite in the Sudan 33 First Aid and Treatment of Snake Bite 34 Index to Snake Names :—• (a) Scientific ... ... ... ... ... ... ... 36 (b) Popular English 37 (c) Vernacular Sudanese 38 Appendix I. List of additional Sudanese Species ... ... 39 Appendix II. Instructions for Collectors 40 3 FOREWORD. HE Sudan Government collection of snakes was first started in 1920, and by 1930 contained 220 specimens. In the latter year T Mr. N. L. Corkill of the Sudan Medical Service, who had recently completed a survey of snakes and snake bite in Iraq, undertook to classify all specimens in the collection and subsequent additions thereto. -

Ancestral Reconstruction of Diet and Fang Condition in the Lamprophiidae: Implications for the Evolution of Venom Systems in Snakes
Journal of Herpetology, Vol. 55, No. 1, 1–10, 2021 Copyright 2021 Society for the Study of Amphibians and Reptiles Ancestral Reconstruction of Diet and Fang Condition in the Lamprophiidae: Implications for the Evolution of Venom Systems in Snakes 1,2 1 1 HIRAL NAIK, MIMMIE M. KGADITSE, AND GRAHAM J. ALEXANDER 1School of Animal, Plant and Environmental Sciences, University of the Witwatersrand, Johannesburg. PO Wits, 2050, Gauteng, South Africa ABSTRACT.—The Colubroidea includes all venomous and some nonvenomous snakes, many of which have extraordinary dental morphology and functional capabilities. It has been proposed that the ancestral condition of the Colubroidea is venomous with tubular fangs. The venom system includes the production of venomous secretions by labial glands in the mouth and usually includes fangs for effective delivery of venom. Despite significant research on the evolution of the venom system in snakes, limited research exists on the driving forces for different fang and dental morphology at a broader phylogenetic scale. We assessed the patterns of fang and dental condition in the Lamprophiidae, a speciose family of advanced snakes within the Colubroidea, and we related fang and dental condition to diet. The Lamprophiidae is the only snake family that includes front-fanged, rear-fanged, and fangless species. We produced an ancestral reconstruction for the family and investigated the pattern of diet and fangs within the clade. We concluded that the ancestral lamprophiid was most likely rear-fanged and that the shift in dental morphology was associated with changes in diet. This pattern indicates that fang loss, and probably venom loss, has occurred multiple times within the Lamprophiidae. -

Amblyodipsas Polylepis (Bocage, 1873) Feeding on the Amphisbaenid Monopeltis Luandae Gans, 1976
Herpetology Notes, volume 14: 205-207 (2021) (published online on 26 January 2021) A snake with an appetite for the rare: Amblyodipsas polylepis (Bocage, 1873) feeding on the amphisbaenid Monopeltis luandae Gans, 1976 Werner Conradie1,* and Pedro Vaz Pinto2,3 Specimens in natural history museum collections catalogue number PEM R22034. The snake measured represent a unique snapshot of the time and place they 634 mm in snout–vent length (no tail length is provided were collected, while the analysis of stomach contents as the tail was truncated). Identification to the nominate often leads to unexpected results and new discoveries. For subspecies A. p. polylepis was based on a series of example, the Angolan lizard Ichnotropis microlepidota characteristics (fide Broadley, 1990), including enlarged Marx, 1956 was described based on material recovered fangs below a small eye; loreal absent; preocular absent; from the crop of a Dark Chanting Goshawk (Melierax one postocular; seven supralabials, with the 3rd and metabates), and the species has not been collected since 4th entering the orbit; seven infralabials, with the first (Marx, 1956; van den Berg, 2018). Specifically, such four in contact with a single pair of genials; temporal an approach is known to provide extremely valuable formula 0+1 on both sides; 19-19-17 midbody scale insights into highly cryptic and rarely sighted fossorial rows; 227 ventrals; 16+ paired subcaudals (truncated). species, such as amphisbaenids (Broadley, 1971; Shine The specimen was re-examined in mid-2019 and it was et al., 2006). These tend to be generally underrepresented discovered that the stomach was full. Upon dissection, a in museum collections and, therefore, make a case for fully intact amphisbaenian was removed (Fig. -

Frogs & Reptiles NE Vic 2018 Online
Reptiles and Frogs of North East Victoria An Identication and Conservation Guide Victorian Conservation Status (DELWP Advisory List) cr critically endangered en endangered Reptiles & Frogs vu vulnerable nt near threatened dd data deficient L Listed under the Flora and Fauna Guarantee Act (FFG, 1988) Size: of North East Victoria Lizards, Dragons & Skinks: Snout-vent length (cm) Snakes, Goannas: Total length (cm) An Identification and Conservation Guide Lowland Copperhead Highland Copperhead Carpet Python Gray's Blind Snake Nobbi Dragon Bearded Dragon Ragged Snake-eyed Skink Large Striped Skink Frogs: Snout-vent length male - M (mm) Snout-vent length female - F (mm) Austrelaps superbus 170 (NC) Austrelaps ramsayi 115 (PR) Morelia spilota metcalfei – en L 240 (DM) Ramphotyphlops nigrescens 38 (PR) Diporiphora nobbi 8.4 (PR) Pogona barbata – vu 25 (DM) Cryptoblepharus pannosus Snout-Vent 3.5 (DM) Ctenotus robustus Snout-Vent 12 (DM) Guide to symbols Venomous Lifeform F Fossorial (burrows underground) T Terrestrial Reptiles & Frogs SA Semi Arboreal R Rock-dwelling Habitat Type Alpine Bog Montane Forests Alpine Grassland/Woodland Lowland Grassland/Woodland White-lipped Snake Tiger Snake Woodland Blind Snake Olive Legless Lizard Mountain Dragon Marbled Gecko Copper-tailed Skink Alpine She-oak Skink Drysdalia coronoides 40 (PR) Notechis scutatus 200 (NC) Ramphotyphlops proximus – nt 50 (DM) Delma inornata 13 (DM) Rankinia diemensis Snout-Vent 7.5 (NC) Christinus marmoratus Snout-Vent 7 (PR) Ctenotus taeniolatus Snout-Vent 8 (DM) Cyclodomorphus praealtus