Phylogenetic Reconstruction of the Narrow-Leaf Cattail Typha Angustifolia L., Using the Chloroplast Maturase K ( Matk ) Gene Sequence
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The 2014 Golden Gate National Parks Bioblitz - Data Management and the Event Species List Achieving a Quality Dataset from a Large Scale Event
National Park Service U.S. Department of the Interior Natural Resource Stewardship and Science The 2014 Golden Gate National Parks BioBlitz - Data Management and the Event Species List Achieving a Quality Dataset from a Large Scale Event Natural Resource Report NPS/GOGA/NRR—2016/1147 ON THIS PAGE Photograph of BioBlitz participants conducting data entry into iNaturalist. Photograph courtesy of the National Park Service. ON THE COVER Photograph of BioBlitz participants collecting aquatic species data in the Presidio of San Francisco. Photograph courtesy of National Park Service. The 2014 Golden Gate National Parks BioBlitz - Data Management and the Event Species List Achieving a Quality Dataset from a Large Scale Event Natural Resource Report NPS/GOGA/NRR—2016/1147 Elizabeth Edson1, Michelle O’Herron1, Alison Forrestel2, Daniel George3 1Golden Gate Parks Conservancy Building 201 Fort Mason San Francisco, CA 94129 2National Park Service. Golden Gate National Recreation Area Fort Cronkhite, Bldg. 1061 Sausalito, CA 94965 3National Park Service. San Francisco Bay Area Network Inventory & Monitoring Program Manager Fort Cronkhite, Bldg. 1063 Sausalito, CA 94965 March 2016 U.S. Department of the Interior National Park Service Natural Resource Stewardship and Science Fort Collins, Colorado The National Park Service, Natural Resource Stewardship and Science office in Fort Collins, Colorado, publishes a range of reports that address natural resource topics. These reports are of interest and applicability to a broad audience in the National Park Service and others in natural resource management, including scientists, conservation and environmental constituencies, and the public. The Natural Resource Report Series is used to disseminate comprehensive information and analysis about natural resources and related topics concerning lands managed by the National Park Service. -

Floristic and Ecological Characterization of Habitat Types on an Inselberg in Minas Gerais, Southeastern Brazil
Acta Botanica Brasilica - 31(2): 199-211. April-June 2017. doi: 10.1590/0102-33062016abb0409 Floristic and ecological characterization of habitat types on an inselberg in Minas Gerais, southeastern Brazil Luiza F. A. de Paula1*, Nara F. O. Mota2, Pedro L. Viana2 and João R. Stehmann3 Received: November 21, 2016 Accepted: March 2, 2017 . ABSTRACT Inselbergs are granitic or gneissic rock outcrops, distributed mainly in tropical and subtropical regions. Th ey are considered terrestrial islands because of their strong spatial and ecological isolation, thus harboring a set of distinct plant communities that diff er from the surrounding matrix. In Brazil, inselbergs scattered in the Atlantic Forest contain unusually high levels of plant species richness and endemism. Th is study aimed to inventory species of vascular plants and to describe the main habitat types found on an inselberg located in the state of Minas Gerais, in southeastern Brazil. A total of 89 species of vascular plants were recorded (belonging to 37 families), of which six were new to science. Th e richest family was Bromeliaceae (10 spp.), followed by Cyperaceae (seven spp.), Orchidaceae and Poaceae (six spp. each). Life forms were distributed in diff erent proportions between habitats, which suggested distinct microenvironments on the inselberg. In general, habitats under similar environmental stress shared common species and life-form proportions. We argue that fl oristic inventories are still necessary for the development of conservation strategies and management of the unique vegetation on inselbergs in Brazil. Keywords: endemism, granitic and gneissic rock outcrops, life forms, terrestrial islands, vascular plants occurring on rock outcrops within the Atlantic Forest Introduction domain, 416 are endemic to these formations (Stehmann et al. -

Vascular Epiphytes of Reserva Ecológica De Guapiaçu
Cachoeiras de Macacu, Atlantic Forest - Rio de Janeiro, Brazil 1 Vascular epiphytes of Reserva Ecológica de Guapiaçu Nadjara de Medeiros Corrêa 1; Mariana Moreira da Silva Murakami 1; Laura do Nascimento Martins 1,2; Ana Carolina Rodrigues da Cruz 1,3 & André Felippe Nunes-Freitas 1 1 Universidade Federal Rural do Rio de Janeiro; 2 Instituto Nacional de Pesquisas da Amazônia; 3 Museu Nacional, Universidade Federal do Rio de Janeiro Photo By: Corrêa N. M.; Cruz, A.C.R.;MurakamiM.M. Produced by: Corrêa N. M. & Juliana Philipp, Keller Science Action Center, Field Museum @Nadjara de Medeiros Corrêa [[email protected]], Mariana Moreira da Silva Murakami, Laura do Nascimento Martins, Ana Carolina Rodrigues da Cruz and André Felippe Nunes-Freitas ©Nadjara de Medeiros Corrêa Support from CAPES; FAPERJ and UFRRJ. [fieldguides.fieldmuseum.org] [1208] version 1 11/2019 1 Anthurium minarum 2 Anthurium scandens 3 Anthurium scandens 4 Anthurium scandens 5 Anthurium solitarium Sakur. & Mayo (Aubl.) Engl. (Aubl.) Engl. (Aubl.) Engl. Schott ARACEAE ARACEAE ARACEAE ARACEAE ARACEAE 6 Anthurium solitarium 7 Monstera adansonii 8 Philodendron cordatum 9 Philodendron cordatum 10 Asplenium serratum L. Schott Schott Schott Schott ASPLENIACEAE ARACEAE ARACEAE ARACEAE ARACEAE 11 Asplenium serratum L. 12 Begonia hirtella Link 13 Begonia hirtella Link 14 Begonia integerrima ASPLENIACEAE BEGONIACEAE BEGONIACEAE Spreng. BEGONIACEAE Cachoeiras de Macacu, Atlantic Forest - Rio de Janeiro, Brazil Vascular epiphytes of Reserva Ecológica de Guapiaçu 2 Nadjara de Medeiros Corrêa 1; Mariana Moreira da Silva Murakami 1; Laura do Nascimento Martins 1,2; Ana Carolina Rodrigues da Cruz 1,3 & André Felippe Nunes-Freitas 1 1 Universidade Federal Rural do Rio de Janeiro; 2 Instituto Nacional de Pesquisas da Amazônia; 3 Museu Nacional, Universidade Federal do Rio de Janeiro Photo By: Corrêa N. -

(BCF), Translocation Factor (TF) and Metal Enrichment Factor (MEF) Abilities of Aquatic Macrophyte Species Exposed to Metal Contaminated Wastewater
ISSN(Online): 2319-8753 ISSN (Print): 2347-6710 International Journal of Innovative Research in Science, Engineering and Technology (A High Impact Factor, Monthly, Peer Reviewed Journal) Visit: www.ijirset.com Vol. 8, Issue 1, January 2019 Evaluation of Bioaccumulation Factor (BAF), Bioconcentration Factor (BCF), Translocation Factor (TF) and Metal Enrichment Factor (MEF) Abilities of Aquatic Macrophyte Species Exposed to Metal Contaminated Wastewater S. S. Shingadgaon1, B.L. Chavan2 Research Scholar, Department of Environmental Science, School of Earth Sciences, Solapur University, Solapur, MS, India1 Former Professor and Head, Department of Environmental Science, Solapur University Solapur and presently working at Department of Environmental Science, Dr.Babasaheb Ambedkar Marathwada University, Aurangabad, MS, India 2 ABSTRACT: Wastewaters receiving aquatic bodies are quiet complex in terms of pollutants, the transport and interactions with heavy metals. This complexity is primarily due to high variability of pollutants, contaminants and related parameters. The macrophytes are plausible bio-indicators of the pollution load and level of metals within the aquatic systems than the wastewater or sediment analyses. The potential ability of aquatic macrophytes in natural water bodies receiving municipal sewage from Solapur city was assessed. Data from the studies on macrophytes exposed to a mixed test bath of metals and examined to know their potentialities to accumulate heavy metals for judging their suitability for phytoremediation technology -

From Genes to Genomes: Botanic Gardens Embracing New Tools for Conservation and Research Volume 18 • Number 1
Journal of Botanic Gardens Conservation International Volume 18 • Number 1 • February 2021 From genes to genomes: botanic gardens embracing new tools for conservation and research Volume 18 • Number 1 IN THIS ISSUE... EDITORS Suzanne Sharrock EDITORIAL: Director of Global Programmes FROM GENES TO GENOMES: BOTANIC GARDENS EMBRACING NEW TOOLS FOR CONSERVATION AND RESEARCH .... 03 Morgan Gostel Research Botanist, FEATURES Fort Worth Botanic Garden Botanical Research Institute of Texas and Director, GGI-Gardens NEWS FROM BGCI .... 06 Jean Linksy FEATURED GARDEN: THE NORTHWESTERN UNIVERSITY Magnolia Consortium Coordinator, ECOLOGICAL PARK & BOTANIC GARDENS .... 09 Atlanta Botanical Garden PLANT HUNTING TALES: GARDENS AND THEIR LESSONS: THE JOURNAL OF A BOTANY STUDENT Farahnoz Khojayori .... 13 Cover Photo: Young and aspiring scientists assist career scientists in sampling plants at the U.S. Botanic Garden for TALKING PLANTS: JONATHAN CODDINGTON, the Global Genome Initiative (U.S. Botanic Garden). DIRECTOR OF THE GLOBAL GENOME INITIATIVE .... 16 Design: Seascape www.seascapedesign.co.uk BGjournal is published by Botanic Gardens Conservation International (BGCI). It is published twice a year. Membership is open to all interested individuals, institutions and organisations that support the aims of BGCI. Further details available from: ARTICLES • Botanic Gardens Conservation International, Descanso House, 199 Kew Road, Richmond, Surrey TW9 3BW UK. Tel: +44 (0)20 8332 5953, Fax: +44 (0)20 8332 5956, E-mail: [email protected], www.bgci.org BANKING BOTANICAL BIODIVERSITY WITH THE GLOBAL GENOME • BGCI (US) Inc, The Huntington Library, BIODIVERSITY NETWORK (GGBN) Art Collections and Botanical Gardens, Ole Seberg, Gabi Dröge, Jonathan Coddington and Katharine Barker .... 19 1151 Oxford Rd, San Marino, CA 91108, USA. -

Entry Worksheet - Bromeliad Society of South Florida
BromeliAdvisory March 2017 InThis Issue President’s Message . 2 Michalski’s Suggestions for Show . 2 The Show is Just Around the Corner . 3-5 In Case You Missed It . 6 Upcoming Events . 6-7 News From Florida Council . 7 Murder in Corbin . 7-8 Renewal of Membership Form . 9 Show Entry Sheet . 10 BromeliAdvisory Stop and Smell the Bromeliads March 2017 MARCH 21, 2017 WEBPAGE: http://www.bssf-miami.org/ SPEAKER: Dave Johnston http://www.facebook.com/groups/BromeliadS RAFFLE TABLE: Barbara Partagas SF/?bookmark_t=group FOOD: Usual Suspects http://www.facebook.com/pages/Bromeliad-S ociety-of-South-Florida/84661684279 Alex Bello, President Dave Johnston Sandy Roth, VP Dave’s interest in Bromeliads began in 1985 when he Fred Sussenberger, Treasurer purchased two bromeliads that Frank’s Lenny Goldstein, Secretary nursery had for sale. A cryptanthus Pink _________________ Starlight and a neoregelia carolinae type. DIRECTORS Soon thereafter, he learned there were Past Pres.: Sandy Roth bromeliad clubs that shared similar Directors: interests. He joined some. By 1990, Barbara Sparling ’16 –’17 Dave had acquired so many bromeliads Karen Bradley ’17 –’18 that he launched Bromeliads Galore – a Ellis Brown’17 –’18 collector’s nursery. Stephanie LaRusso ‘17-‘18 Over the past 32 years, Dave has Editor: Robert Meyer acquired over 2,000 different species and hybrids. As time passes, he became more sensible and has since significantly scaled back the number of plants – now he Advertising: Robert Meyer has perhaps 1200 different species or hybrids. Door Prize: Alan Herndon Education: Alan Herndon Dave has served five terms as President of The Bromeliad Hospitality: Elaine Mills Guild of Tampa and two terms as President of the Florida Library: Barbara Partagas West Coast Bromeliad Society. -

The Florida East Coast Bromeliad Society June 2014
1 The Florida East Coast Bromeliad Society June 2014 The Florida East Coast Bromeliad Society Next meeting Sunday, June 8th, 2014 1:30pm. Colony in the Wood – club house 4000 S. Clyde Morris Blvd., Port Orange 32129 June 2014 Summertime is the Right Time… President – Jay Thurrott 386/761-4804 Vice President – Bill Hazard 386/882-3850 Secretary – Jillian Ragia 386/801-2500 Treasurer – Eve Krauth 386/763-2084 ________________________________________________________________________________ When is a good time to separate bromeliad to do it, but somehow Tree managed to pups? When should we repot existing prepare and orchestrate placement of many bromeliads – either to a larger pot size or just of the refreshments for the meeting while to replace the “worn-out” potting mix? When visiting with family for Mothers Day at the are the oak tree leaves finally going to stop same time as the meeting. That’s an example falling into my bromeliads so I can clean all of ‘multi-tasking’ at its best and our sincerest of the fallen debris out of the leaf axils? That thanks go Tree and Bill for their hard work in time is here! It’s summertime and if you plan hosting our May FECBS meeting! Everyone your workdays in the garden so that you can “ooh’d” and “aaaah’d” over the use of avoid the mid-day heat of Florida’s summer, bromeliads in the landscaping and many you can accomplish a lot in the garden. went home with plants that Bill had so Summer in Northeast Florida is when our generously donated for the raffle. -

FLORIDA WEST COAST BROMELIAD SOCIETY 1954-2014 Celebrating 60 Years of Bromeliads Floridabromeliads.Org
FLORIDA WEST COAST BROMELIAD SOCIETY 1954-2014 Celebrating 60 Years of Bromeliads floridabromeliads.org July 2014 Newsletter NEXT MEETING Date & Time: Location: Tuesday, July 1 Good Samaritan Church Doors open at 7 pm; meeting starts at 7:30 6085 Park Boulevard Pinellas Park, Florida 33781 Program Dennis Cathcart’s presentation, New Explorations and Discoveries in Brazil--Part I, is based on the recent five-week trip he and his wife Linda took to Brazil that took them through four of Brazil’s 26 states. Their objectives were to find Dyckias and other terrestrial bromeliads and visit some of the bromeliad colonies they had known from past visits to Brazil. Along the way they found the Dyckias and so much more—varied scenery, diverse biological zones and climates, wildlife, insects, and plants. Hopefully the ‘Part I’ in the title means we can look forward to additional chapters in their 2014 Brazil tour story. Dennis and Linda have owned and operated Tropiflora Nursery in Sarasota, Florida, since 1976. The nursery specializes in rare and exotic plants, primarily bromeliads along with orchids, cycads, caudiciforms, succulents, ferns, palms and more. It is one of the largest collector-oriented exotic plant nurseries in the United States and has over 200,000 square feet of greenhouses. Many of the plants in their collection were collected by them as seeds and cuttings on their more than100 collecting expeditions made in over 35 years and in 27 countries. Plant Sales The speaker will be the sole plant vendor for this meeting and there will be no member plant sales. -
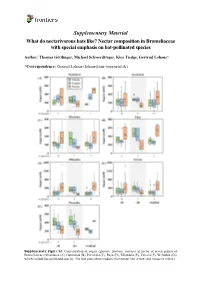
Supplementary Material What Do Nectarivorous Bats Like? Nectar Composition in Bromeliaceae with Special Emphasis on Bat-Pollinated Species
Supplementary Material What do nectarivorous bats like? Nectar composition in Bromeliaceae with special emphasis on bat-pollinated species Author: Thomas Göttlinger, Michael Schwerdtfeger, Kira Tiedge, Gertrud Lohaus* *Correspondence: Gertrud Lohaus ([email protected]) Supplementary Figure S1: Concentration of sugars (glucose, fructose, sucrose) in nectar of seven genera of Bromeliaceae (Alcantarea (A), Guzmania (B), Pitcairnia (C), Puya (D), Tillandsia (E), Vriesea (F), Werauhia (G)) which include bat-pollinated species. The box plots show medians (horizontal line in box) and means (x in box). Supplementary Material What do nectarivorous bats like? Nectar composition in Bromeliaceae with special emphasis on bat-pollinated species Author: Thomas Göttlinger, Michael Schwerdtfeger, Kira Tiedge, Gertrud Lohaus* *Correspondence: Gertrud Lohaus ([email protected]) Supplementary Figure S2: Concentration of amino acids (ala, arg, asn, asp, gaba, gln, glu, gly, his, iso, leu, lys, met, phe, pro, ser, thr, trp, tyr, val) in nectar of seven genera of Bromeliaceae (Alcantarea (A), Guzmania (B), Pitcairnia (C), Puya (D), Tillandsia (E), Vriesea (F), Werauhia (G)), which include bat-pollinated species. The box plots show medians (horizontal line in box) and means (x in box). Supplementary Material What do nectarivorous bats like? Nectar composition in Bromeliaceae with special emphasis on bat-pollinated species Author: Thomas Göttlinger, Michael Schwerdtfeger, Kira Tiedge, Gertrud Lohaus* *Correspondence: Gertrud Lohaus ([email protected]) Supplementary Figure S3: Cation concentrations (Ca2+, K+, Na+, Mg2+) in nectar of seven genera of Bromeliaceae (Alcantarea (A), Guzmania (B), Pitcairnia (C), Puya (D), Tillandsia (E), Vriesea (F), Werauhia (G)), which include bat-pollinated species. The box plots show medians (horizontal line in box) and means (x in box). -

VARIEGATION in BROMELIADS (By Luiz Felipe Nevares De Carvalho)
VARIEGATION IN BROMELIADS (by Luiz Felipe Nevares de Carvalho) Editorial comment (Bob Reilly) Reprinted, with permission of the Bromeliad Society International, from the Journal of The Bromeliad Society, 2000, volume 50 (4), pp. 182-185. Variegated bromeliads are often keenly sought after by collectors. In this article, the author discusses the causes and types of variegation, as well as the propagation of variegated bromeliads. Note that the process of naming a particular variegated plant can sometimes be more complex than might be inferred from the article. Variegation is a rather common phenomenon in the plant kingdom, and is found in many plant families. It is especially pronounced in Bromeliaceae. The word “variegata” comes from Latin – variegatuus, variegata, variegatum – meaning variable coloration with patches of different colors. A bromeliad is known as “variegata” when it has two or more different colors. Over 60% of cultivated bromeliads have bands, dots, lines, and streaks, and can therefore be considered variegated. However, the term is accepted in horticulture, when applied to bromeliads that have lines, streaks and longitudinal bands of contrasting colors, especially those that show differences in pigmentation between the green chlorophyll-containing tissues and albino tissues. On the other hand, if we look at the many bromeliads that grow in the wild, it appears that variegation is a rare phenomenon. As a general rule, patently variegated plants are less hardy and slower growing than normal, and those that arise spontaneously in nature normally survive the competition for space and light only when man intervenes, taking them from the wild for cultivation. Variegation is rarely found in the subfamily Pitcairnioideae, and is not particularly common in Tillandsioideae. -

Redalyc.Plant-Hummingbird Interactions and Temporal Nectar
Anais da Academia Brasileira de Ciências ISSN: 0001-3765 [email protected] Academia Brasileira de Ciências Brasil FONSECA, LORENA C.N.; VIZENTIN-BUGONI, JEFERSON; RECH, ANDRÉ R.; ALVES, MARIA ALICE S. Plant-hummingbird interactions and temporal nectar availability in a restinga from Brazil Anais da Academia Brasileira de Ciências, vol. 87, núm. 4, octubre-diciembre, 2015, pp. 2163-2175 Academia Brasileira de Ciências Rio de Janeiro, Brasil Available in: http://www.redalyc.org/articulo.oa?id=32743236022 How to cite Complete issue Scientific Information System More information about this article Network of Scientific Journals from Latin America, the Caribbean, Spain and Portugal Journal's homepage in redalyc.org Non-profit academic project, developed under the open access initiative Anais da Academia Brasileira de Ciências (2015) 87(4): 2163-2175 (Annals of the Brazilian Academy of Sciences) Printed version ISSN 0001-3765 / Online version ISSN 1678-2690 http://dx.doi.org/10.1590/0001-3765201520140349 www.scielo.br/aabc Plant-hummingbird interactions and temporal nectar availability in a restinga from Brazil LORENA C.N. FONSECA1,5, JEFERSON VIZENTIN-BUGONI2,6, ANDRÉ R. RECH3 and MARIA ALICE S. ALVES4 1Programa de Pós-Graduação em Ecologia, Universidade Federal do Rio de Janeiro/ UFRJ, CCS, IB, Caixa Postal 68020, 21941-590 Rio de Janeiro, RJ, Brasil 2Programa de Pós-Graduação em Ecologia, Instituto de Biologia, Universidade Estadual de Campinas/ UNICAMP, Rua Monteiro Lobato, 970, Barão Geraldo, 13083-970 Campinas, SP, Brasil 3Instituto de Biologia, Universidade Federal de Uberlândia/UFU, Av. Pará, 1720, Campus Umuarama, 38405-320 Uberlândia, MG, Brasil 4Departamento de Ecologia, Instituto de Biologia Roberto Alcantara Gomes, Universidade do Estado do Rio de Janeiro/UERJ, Rua São Francisco Xavier, 524, Maracanã, 20550-011 Rio de Janeiro, RJ, Brasil 5Companhia Ambiental do Estado de São Paulo/CETESB, Av. -

Plant-Hummingbird Interactions and Temporal Nectar Availability in a Restinga from Brazil
Anais da Academia Brasileira de Ciências (2015) 87(4): 2163-2175 (Annals of the Brazilian Academy of Sciences) Printed version ISSN 0001-3765 / Online version ISSN 1678-2690 http://dx.doi.org/10.1590/0001-3765201520140349 www.scielo.br/aabc Plant-hummingbird interactions and temporal nectar availability in a restinga from Brazil LORENA C.N. FONSECA1,5, JEFERSON VIZENTIN-BUGONI2,6, ANDRÉ R. RECH3 and MARIA ALICE S. ALVES4 1Programa de Pós-Graduação em Ecologia, Universidade Federal do Rio de Janeiro/ UFRJ, CCS, IB, Caixa Postal 68020, 21941-590 Rio de Janeiro, RJ, Brasil 2Programa de Pós-Graduação em Ecologia, Instituto de Biologia, Universidade Estadual de Campinas/ UNICAMP, Rua Monteiro Lobato, 970, Barão Geraldo, 13083-970 Campinas, SP, Brasil 3Instituto de Biologia, Universidade Federal de Uberlândia/UFU, Av. Pará, 1720, Campus Umuarama, 38405-320 Uberlândia, MG, Brasil 4Departamento de Ecologia, Instituto de Biologia Roberto Alcantara Gomes, Universidade do Estado do Rio de Janeiro/UERJ, Rua São Francisco Xavier, 524, Maracanã, 20550-011 Rio de Janeiro, RJ, Brasil 5Companhia Ambiental do Estado de São Paulo/CETESB, Av. Professor Frederico Hermann Jr, 345, Pinheiros, 05459-900 São Paulo, SP, Brasil 6Center for Macroecology, Evolution and Climate, Natural History Museum of Denmark, University of Copenhagen, Universitetsparken 15, DK-2100 Copenhagen Ø, Denmark Manuscript received on July 10, 2014; accepted for publication on April 27, 2015 ABSTRACT Hummingbirds are the most important and specialized group of pollinating birds in the Neotropics and their interactions with plants are key components to many communities. In the present study we identified the assemblage of plants visited by hummingbirds and investigated the temporal availability of floral resources in an area of restinga, sandy plain coastal vegetation associated with the Atlantic forest, in Southeastern Brazil.