Endolysosomal Degradation of Tau and Its Role in Glucocorticoid-Driven Hippocampal
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

82755929.Pdf
FEBS Letters 589 (2015) 2931–2943 journal homepage: www.FEBSLetters.org Review The 4D nucleome: Evidence for a dynamic nuclear landscape based on co-aligned active and inactive nuclear compartments ⇑ Thomas Cremer a, , Marion Cremer a, Barbara Hübner a,1, Hilmar Strickfaden b, Daniel Smeets a, ⇑ ⇑ Jens Popken a, Michael Sterr a,2, Yolanda Markaki a, Karsten Rippe c, , Christoph Cremer d, a Biocenter, Department Biology II, Ludwig Maximilians University (LMU), Martinsried, Germany b University of Alberta, Cross Cancer Institute Dept. of Oncology, Edmonton, AB, Canada c German Cancer Research Center (DKFZ) & BioQuant Center, Research Group Genome Organization & Function, Heidelberg, Germany d Institute of Molecular Biology (IMB), Mainz and Institute of Pharmacy and Molecular Biotechnology (IPMB), University of Heidelberg, Germany article info abstract Article history: Recent methodological advancements in microscopy and DNA sequencing-based methods provide Received 1 April 2015 unprecedented new insights into the spatio-temporal relationships between chromatin and nuclear Revised 19 May 2015 machineries. We discuss a model of the underlying functional nuclear organization derived mostly Accepted 20 May 2015 from electron and super-resolved fluorescence microscopy studies. It is based on two spatially Available online 28 May 2015 co-aligned, active and inactive nuclear compartments (ANC and INC). The INC comprises the com- Edited by Wilhelm Just pact, transcriptionally inactive core of chromatin domain clusters (CDCs). The ANC is formed by the transcriptionally active periphery of CDCs, called the perichromatin region (PR), and the inter- chromatin compartment (IC). The IC is connected to nuclear pores and serves nuclear import and Keywords: 4D nucleome export functions. The ANC is the major site of RNA synthesis. -

The Elucidation of Stress Memory Inheritance in Brassica Rapa Plants
ORIGINAL RESEARCH ARTICLE published: 21 January 2015 doi: 10.3389/fpls.2015.00005 The elucidation of stress memory inheritance in Brassica rapa plants Andriy Bilichak 1, Yaroslav Ilnytskyy 2, Rafal Wóycicki 2, Nina Kepeshchuk 2,DawsonFogen2 and Igor Kovalchuk 2* 1 Lethbridge Research Centre, Agriculture and Agri-Food Canada, Lethbridge, AB, Canada 2 Department of Biological Sciences, University of Lethbridge, Lethbridge, AB, Canada Edited by: Plants are able to maintain the memory of stress exposure throughout their ontogenesis Shawn Kaeppler, University of and faithfully propagate it into the next generation. Recent evidence argues for the Wisconsin-Madison, USA epigenetic nature of this phenomenon. Small RNAs (smRNAs) are one of the vital Reviewed by: epigenetic factors because they can both affect gene expression at the place of Rajandeep Sekhon, Clemson University, USA their generation and maintain non-cell-autonomous gene regulation. Here, we have Thelma Farai Madzima, Florida State made an attempt to decipher the contribution of smRNAs to the heat-shock-induced University, USA transgenerational inheritance in Brassica rapa plants using sequencing technology. To *Correspondence: do this, we have generated comprehensive profiles of a transcriptome and a small Igor Kovalchuk, Department of RNAome (smRNAome) from somatic and reproductive tissues of stressed plants and their Biological Sciences, University of Lethbridge, University Drive 4401, untreated progeny. We have demonstrated that the highest tissue-specific alterations in Lethbridge, AB, T1K 3M4, Canada the transcriptome and smRNAome profile are detected in tissues that were not directly e-mail: [email protected] exposed to stress, namely, in the endosperm and pollen. Importantly, we have revealed that the progeny of stressed plants exhibit the highest fluctuations at the smRNAome level but not at the transcriptome level. -

Warning Sines: Alu Elements, Evolution of the Human Brain, and the Spectrum of Neurological Disease
Chromosome Res (2018) 26:93–111 https://doi.org/10.1007/s10577-018-9573-4 ORIGINAL ARTICLE Warning SINEs: Alu elements, evolution of the human brain, and the spectrum of neurological disease Peter A. Larsen & Kelsie E. Hunnicutt & Roxanne J. Larsen & Anne D. Yoder & Ann M. Saunders Received: 6 December 2017 /Revised: 14 January 2018 /Accepted: 15 January 2018 /Published online: 19 February 2018 # The Author(s) 2018. This article is an open access publication Abstract Alu elements are a highly successful family of neurological networks are potentially vulnerable to the primate-specific retrotransposons that have fundamen- epigenetic dysregulation of Alu elements operating tally shaped primate evolution, including the evolution across the suite of nuclear-encoded mitochondrial genes of our own species. Alus play critical roles in the forma- that are critical for both mitochondrial and CNS func- tion of neurological networks and the epigenetic regu- tion. Here, we highlight the beneficial neurological as- lation of biochemical processes throughout the central pects of Alu elements as well as their potential to cause nervous system (CNS), and thus are hypothesized to disease by disrupting key cellular processes across the have contributed to the origin of human cognition. De- CNS. We identify at least 37 neurological and neurode- spite the benefits that Alus provide, deleterious Alu generative disorders wherein deleterious Alu activity has activity is associated with a number of neurological been implicated as a contributing factor for the manifes- and neurodegenerative disorders. In particular, tation of disease, and for many of these disorders, this activity is operating on genes that are essential for proper mitochondrial function. -

Family Name Files That Can Be Accessed in the Boerne Public Library Local and Family History Archives
FAMILY NAME FILES THAT CAN BE ACCESSED IN THE BOERNE PUBLIC LIBRARY LOCAL AND FAMILY HISTORY ARCHIVES A Abadie, Ables, Abshire Ackerman Adair, Adam, Adamek, Adamietz, Adams, Adamson, Adcock, Adkins, Adler, Adrian, Adriance, Agold, Aguirre, Ahr, Ahrlett, Alba, Albrecht, Albright, Alcorn, Alderman, Aldridge, Alexander, Alf, Alford, Algueseva, Allamon, Allen, Allison, Altgelt, Alvarez, Amason, Ammann, Ames, Anchondo, Anderson, Angell, Angle, Ansaldo, Anstiss, Appelt, Armendarez, Armstrong, Arnold, Arnott, Artz, Ashcroft, Asher, Atencio, Atkinson, Aue, Austin, Aycock, B Backer, Bacon, Bacorn, Bain, Baker, Baldwin, Ball, Balser, Bangert, Bankier, Banks, Bannister, Barbaree, Barker, Barkley, Barnard, Barnes, Barnett, Barnette, Barnhart, Baron, Barrett, Barrington, Barron, Barrows, Barta, Barteau, Bartel, Bartels, Barthlow, Barton, Basham, Bates, Batha, Bauer, Baum, Baumann, Bausch, Baxter, Bayard, Bayless, Baynton, Beagle, Bealor, Beam, Bean, Beardslee, Beasley, Beath, Beatty, Beauford, Beaver, Bechtold, Beck, Becker, Beckett, Beckley, Bedford, Bedgood, Bednar, Bedwell, Beem, Beene, Beer, Begia, Behr, Beissner, Bell, Below, Bemus, Benavides, Bender, Bennack, Benefield, Benner, Bennett, Benson, Bentley, Berger, Bergmann, Berlin, Berline, Bernal, Berne, Bernert, Bernhard, Bernstein, Berry, Besch, Beseler, Beshea, Besser, Best, Bettison, Beutnagel, Bevers, Bey, Beyer, Bickel, Bidus, Bien, Bierman, Bierschwale, Bigger, Biggs, Billingsley, Birdsong, Birkner, Bish, Bitzkie, Black, Blackburn, Blackford, Blackwell, Blair, Blaize, Blake, Blakey, Blalock, -

Germanic Surname Lexikon
Germanic Surname Lexikon Most Popular German Last Names with English Meanings German for Beginners: Contents - Free online German course Introduction German Vocabulary: English-German Glossaries For each Germanic surname in this databank we have provided the English Free Online Translation To or From German meaning, which may or may not be a surname in English. This is not a list of German for Beginners: equivalent names, but rather a sampling of English translations of German names. Das Abc German for Beginners: In many cases, there may be several possible origins or translations for a Lektion 1 surname. The translation shown for a surname may not be the only possibility. Some names are derived from Old German and may have a different meaning from What's Hot that in modern German. Name research is not always an exact science. German Word of the Day - 25. August German Glossary of Abbreviations: OHG (Old High German) Words to Avoid - T German Words to Avoid - Glossary Warning German Glossary of Germanic Last Names (A-K) Words to Avoid - With English Meanings Feindbilder German Words to Avoid - Nachname Last Name English Meaning A Special Glossary Close Ad A Dictionary of German Aachen/Aix-la-Chapelle Names Aachen/Achen (German city) Hans Bahlow, Edda ... Best Price $16.95 Abend/Abendroth evening/dusk or Buy New $19.90 Abt abbott Privacy Information Ackerman(n) farmer Adler eagle Amsel blackbird B Finding Your German Ancestors Bach brook Kevan M Hansen Best Price $4.40 Bachmeier farmer by the brook or Buy New $9.13 Bader/Baader bath, spa keeper Privacy Information Baecker/Becker baker Baer/Bar bear Barth beard Bauer farmer, peasant In Search of Your German Roots. -

Surname Folders.Pdf
SURNAME Where Filed Aaron Filed under "A" Misc folder Andrick Abdon Filed under "A" Misc folder Angeny Abel Anger Filed under "A" Misc folder Aberts Angst Filed under "A" Misc folder Abram Angstadt Achey Ankrum Filed under "A" Misc folder Acker Anns Ackerman Annveg Filed under “A” Misc folder Adair Ansel Adam Anspach Adams Anthony Addleman Appenzeller Ader Filed under "A" Misc folder Apple/Appel Adkins Filed under "A" Misc folder Applebach Aduddell Filed under “A” Misc folder Appleman Aeder Appler Ainsworth Apps/Upps Filed under "A" Misc folder Aitken Filed under "A" Misc folder Apt Filed under "A" Misc folder Akers Filed under "A" Misc folder Arbogast Filed under "A" Misc folder Albaugh Filed under "A" Misc folder Archer Filed under "A" Misc folder Alberson Filed under “A” Misc folder Arment Albert Armentrout Albight/Albrecht Armistead Alcorn Armitradinge Alden Filed under "A" Misc folder Armour Alderfer Armstrong Alexander Arndt Alger Arnold Allebach Arnsberger Filed under "A" Misc folder Alleman Arrel Filed under "A" Misc folder Allen Arritt/Erret Filed under “A” Misc folder Allender Filed under "A" Misc folder Aschliman/Ashelman Allgyer Ash Filed under “A” Misc folder Allison Ashenfelter Filed under "A" Misc folder Allumbaugh Filed under "A" Misc folder Ashoff Alspach Asper Filed under "A" Misc folder Alstadt Aspinwall Filed under "A" Misc folder Alt Aston Filed under "A" Misc folder Alter Atiyeh Filed under "A" Misc folder Althaus Atkins Filed under "A" Misc folder Altland Atkinson Alwine Atticks Amalong Atwell Amborn Filed under -
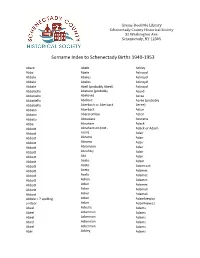
Surname Index to Schenectady Births 1940-1953
Grems-Doolittle Library Schenectady County Historical Society 32 Washington Ave. Schenectady, NY 12305 Surname Index to Schenectady Births 1940-1953 Abare Abele Ackley Abba Abele Ackroyd Abbale Abeles Ackroyd Abbale Abeles Ackroyd Abbale Abell (probably Abeel) Ackroyd Abbatiello Abelone (probably Acord Abbatiello Abelove) Acree Abbatiello Abelove Acree (probably Abbatiello Aberbach or Aberback Aeree) Abbato Aberback Acton Abbato Abercrombie Acton Abbato Aboudara Acucena Abbe Abraham Adack Abbott Abrahamson (not - Adack or Adach Abbott nson) Adair Abbott Abrams Adair Abbott Abrams Adair Abbott Abramson Adair Abbott Abrofsky Adair Abbott Abt Adair Abbott Aceto Adam Abbott Aceto Adamczak Abbott Aceto Adamec Abbott Aceto Adamec Abbott Acken Adamec Abbott Acker Adamec Abbott Acker Adamek Abbott Acker Adamek Abbzle = ? spelling Acker Adamkiewicz unclear Acker Adamkiewicz Abeel Ackerle Adams Abeel Ackerman Adams Abeel Ackerman Adams Abeel Ackerman Adams Abeel Ackerman Adams Abel Ackley Adams Grems-Doolittle Library Schenectady County Historical Society 32 Washington Ave. Schenectady, NY 12305 Surname Index to Schenectady Births 1940-1953 Adams Adamson Ahl Adams Adanti Ahles Adams Addis Ahman Adams Ademec or Adamec Ahnert Adams Adinolfi Ahren Adams Adinolfi Ahren Adams Adinolfi Ahrendtsen Adams Adinolfi Ahrendtsen Adams Adkins Ahrens Adams Adkins Ahrens Adams Adriance Ahrens Adams Adsit Aiken Adams Aeree Aiken Adams Aernecke Ailes = ? Adams Agans Ainsworth Adams Agans Aker (or Aeher = ?) Adams Aganz (Agans ?) Akers Adams Agare or Abare = ? Akerson Adams Agat Akin Adams Agat Akins Adams Agen Akins Adams Aggen Akland Adams Aggen Albanese Adams Aggen Alberding Adams Aggen Albert Adams Agnew Albert Adams Agnew Albert or Alberti Adams Agnew Alberti Adams Agostara Alberti Adams Agostara (not Agostra) Alberts Adamski Agree Albig Adamski Ahave ? = totally Albig Adamson unclear Albohm Adamson Ahern Albohm Adamson Ahl Albohm (not Albolm) Adamson Ahl Albrezzi Grems-Doolittle Library Schenectady County Historical Society 32 Washington Ave. -
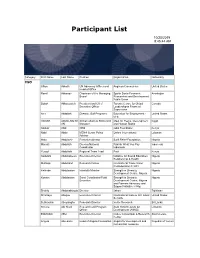
Participant List
Participant List 10/20/2019 8:45:44 AM Category First Name Last Name Position Organization Nationality CSO Jillian Abballe UN Advocacy Officer and Anglican Communion United States Head of Office Ramil Abbasov Chariman of the Managing Spektr Socio-Economic Azerbaijan Board Researches and Development Public Union Babak Abbaszadeh President and Chief Toronto Centre for Global Canada Executive Officer Leadership in Financial Supervision Amr Abdallah Director, Gulf Programs Educaiton for Employment - United States EFE HAGAR ABDELRAHM African affairs & SDGs Unit Maat for Peace, Development Egypt AN Manager and Human Rights Abukar Abdi CEO Juba Foundation Kenya Nabil Abdo MENA Senior Policy Oxfam International Lebanon Advisor Mala Abdulaziz Executive director Swift Relief Foundation Nigeria Maryati Abdullah Director/National Publish What You Pay Indonesia Coordinator Indonesia Yussuf Abdullahi Regional Team Lead Pact Kenya Abdulahi Abdulraheem Executive Director Initiative for Sound Education Nigeria Relationship & Health Muttaqa Abdulra'uf Research Fellow International Trade Union Nigeria Confederation (ITUC) Kehinde Abdulsalam Interfaith Minister Strength in Diversity Nigeria Development Centre, Nigeria Kassim Abdulsalam Zonal Coordinator/Field Strength in Diversity Nigeria Executive Development Centre, Nigeria and Farmers Advocacy and Support Initiative in Nig Shahlo Abdunabizoda Director Jahon Tajikistan Shontaye Abegaz Executive Director International Insitute for Human United States Security Subhashini Abeysinghe Research Director Verite -

Muslims in Spain, 1492–1814 Mediterranean Reconfigurations Intercultural Trade, Commercial Litigation, and Legal Pluralism
Muslims in Spain, 1492– 1814 Mediterranean Reconfigurations Intercultural Trade, Commercial Litigation, and Legal Pluralism Series Editors Wolfgang Kaiser (Université Paris I, Panthéon- Sorbonne) Guillaume Calafat (Université Paris I, Panthéon- Sorbonne) volume 3 The titles published in this series are listed at brill.com/ cmed Muslims in Spain, 1492– 1814 Living and Negotiating in the Land of the Infidel By Eloy Martín Corrales Translated by Consuelo López- Morillas LEIDEN | BOSTON This is an open access title distributed under the terms of the CC BY-NC 4.0 license, which permits any non-commercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited. Further information and the complete license text can be found at https://creativecommons.org/licenses/by-nc/4.0/ The terms of the CC license apply only to the original material. The use of material from other sources (indicated by a reference) such as diagrams, illustrations, photos and text samples may require further permission from the respective copyright holder. Cover illustration: “El embajador de Marruecos” (Catalog Number: G002789) Museo del Prado. Library of Congress Cataloging-in-Publication Data Names: Martín Corrales, E. (Eloy), author. | Lopez-Morillas, Consuelo, translator. Title: Muslims in Spain, 1492-1814 : living and negotiating in the land of the infidel / by Eloy Martín-Corrales ; translated by Consuelo López-Morillas. Description: Leiden ; Boston : Brill, [2021] | Series: Mediterranean reconfigurations ; volume 3 | Original title unknown. | Includes bibliographical references and index. Identifiers: LCCN 2020046144 (print) | LCCN 2020046145 (ebook) | ISBN 9789004381476 (hardback) | ISBN 9789004443761 (ebook) Subjects: LCSH: Muslims—Spain—History. | Spain—Ethnic relations—History. -

Surnames in Bureau of Catholic Indian
RAYNOR MEMORIAL LIBRARIES Montana (MT): Boxes 13-19 (4,928 entries from 11 of 11 schools) New Mexico (NM): Boxes 19-22 (1,603 entries from 6 of 8 schools) North Dakota (ND): Boxes 22-23 (521 entries from 4 of 4 schools) Oklahoma (OK): Boxes 23-26 (3,061 entries from 19 of 20 schools) Oregon (OR): Box 26 (90 entries from 2 of - schools) South Dakota (SD): Boxes 26-29 (2,917 entries from Bureau of Catholic Indian Missions Records 4 of 4 schools) Series 2-1 School Records Washington (WA): Boxes 30-31 (1,251 entries from 5 of - schools) SURNAME MASTER INDEX Wisconsin (WI): Boxes 31-37 (2,365 entries from 8 Over 25,000 surname entries from the BCIM series 2-1 school of 8 schools) attendance records in 15 states, 1890s-1970s Wyoming (WY): Boxes 37-38 (361 entries from 1 of Last updated April 1, 2015 1 school) INTRODUCTION|A|B|C|D|E|F|G|H|I|J|K|L|M|N|O|P|Q|R|S|T|U| Tribes/ Ethnic Groups V|W|X|Y|Z Library of Congress subject headings supplemented by terms from Ethnologue (an online global language database) plus “Unidentified” and “Non-Native.” INTRODUCTION This alphabetized list of surnames includes all Achomawi (5 entries); used for = Pitt River; related spelling vartiations, the tribes/ethnicities noted, the states broad term also used = California where the schools were located, and box numbers of the Acoma (16 entries); related broad term also used = original records. Each entry provides a distinct surname Pueblo variation with one associated tribe/ethnicity, state, and box Apache (464 entries) number, which is repeated as needed for surname Arapaho (281 entries); used for = Arapahoe combinations with multiple spelling variations, ethnic Arikara (18 entries) associations and/or box numbers. -

Iron, Steel and Swords Script - Page 1 Steel Steel, with Plenty of Slag Inclusions
10.3.2 The Iron Trade The town I grew up in after 1950 had around 1000 inhabitants. It was essentially a "sleeping town" for the workers and employes of the factories and businesses in the near-by cities but also still a community of farmers and artisans. Of course there was black smith (and a cartwright, glazier, carpenter, ...), and he was working right across the street from my parent's house. Same thing in all the other towns with a farming background. My grandfather actually was the black smith in another small town with less than 1000 inhabitants. So was his father and grandfather. I'm absolutely sure that any community with more than 500 inhabitants or so supported and needed a black smith ("Schmied" in German) for about 2000 years until - roughly - 1960. The surname Schmid / Schmidt / Schmitt / Schmit is No. 2 on the list of the most frequent surnames in Germany, only bested by "Müller" (you guessed it). It is clear that all those smiths' could not make their own iron / steel but bought it. The major source was the next local smelter "facility" of course, but some long-distance trade with special merchandise also existed. As soon as people started to make iron in quantities, they also started to trade the stuff. The roots of the iron trade thus go back to distant antiquity; we just don't know all that much about it. As we have learned in the preceding parts, a smelter makes a bloom, and this bloom should be processed immediately into one or several pieces of iron that can then be used for making products. -

Surname First Name Categorisation Abadin Jose Luis Silver Abbelen
2018 DRIVERS' CATEGORISATION LIST Updated on 09/07/2018 Drivers in red : revised categorisation Drivers in blue : new categorisation Surname First name Categorisation Abadin Jose Luis Silver Abbelen Klaus Bronze Abbott Hunter Silver Abbott James Silver Abe Kenji Bronze Abelli Julien Silver Abergel Gabriele Bronze Abkhazava Shota Bronze Abra Richard Silver Abreu Attila Gold Abril Vincent Gold Abt Christian Silver Abt Daniel Gold Accary Thomas Silver Acosta Hinojosa Julio Sebastian Silver Adam Jonathan Platinum Adams Rudi Bronze Adorf Dirk Silver Aeberhard Juerg Silver Afanasiev Sergei Silver Agostini Riccardo Gold Aguas Rui Gold Ahlin-Kottulinsky Mikaela Silver Ahrabian Darius Bronze Ajlani Karim Bronze Akata Emin Bronze Aksenov Stanislas Silver Al Faisal Abdulaziz Silver Al Harthy Ahmad Silver Al Masaood Humaid Bronze Al Qubaisi Khaled Bronze Al-Azhari Karim Bronze Alberico Neil Silver Albers Christijan Platinum Albert Michael Silver Albuquerque Filipe Platinum Alder Brian Silver Aleshin Mikhail Platinum Alesi Giuliano Silver Alessi Diego Silver Alexander Iradj Silver Alfaisal Saud Bronze Alguersuari Jaime Platinum Allegretta Vincent Silver Alleman Cyndie Silver Allemann Daniel Bronze Allen James Silver Allgàuer Egon Bronze Allison Austin Bronze Allmendinger AJ Gold Allos Manhal Bronze Almehairi Saeed Silver Almond Michael Silver Almudhaf Khaled Bronze Alon Robert Silver Alonso Fernando Platinum Altenburg Jeff Bronze Altevogt Peter Bronze Al-Thani Abdulrahman Silver Altoè Giacomo Silver Aluko Kolawole Bronze Alvarez Juan Cruz Silver Alzen