Neurodegeneration Induces a Developmental RNA Processing Program by Calpain
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

High Constitutive Cytokine Release by Primary Human Acute Myeloid Leukemia Cells Is Associated with a Specific Intercellular Communication Phenotype
Supplementary Information High Constitutive Cytokine Release by Primary Human Acute Myeloid Leukemia Cells Is Associated with a Specific Intercellular Communication Phenotype Håkon Reikvam 1,2,*, Elise Aasebø 1, Annette K. Brenner 2, Sushma Bartaula-Brevik 1, Ida Sofie Grønningsæter 2, Rakel Brendsdal Forthun 2, Randi Hovland 3,4 and Øystein Bruserud 1,2 1 Department of Clinical Science, University of Bergen, 5020, Bergen, Norway 2 Department of Medicine, Haukeland University Hospital, 5021, Bergen, Norway 3 Department of Medical Genetics, Haukeland University Hospital, 5021, Bergen, Norway 4 Institute of Biomedicine, University of Bergen, 5020, Bergen, Norway * Correspondence: [email protected]; Tel.: +55-97-50-00 J. Clin. Med. 2019, 8, x 2 of 36 Figure S1. Mutational studies in a cohort of 71 AML patients. The figure shows the number of patients with the various mutations (upper), the number of mutations in for each patient (middle) and the number of main classes with mutation(s) in each patient (lower). 2 J. Clin. Med. 2019, 8, x; doi: www.mdpi.com/journal/jcm J. Clin. Med. 2019, 8, x 3 of 36 Figure S2. The immunophenotype of primary human AML cells derived from 62 unselected patients. The expression of the eight differentiation markers CD13, CD14, CD15, CD33, CD34, CD45, CD117 and HLA-DR was investigated for 62 of the 71 patients included in our present study. We performed an unsupervised hierarchical cluster analysis and identified four patient main clusters/patient subsets. The mutational profile for each f the 62 patients is also given (middle), no individual mutation of main class of mutations showed any significant association with any of the for differentiation marker clusters (middle). -
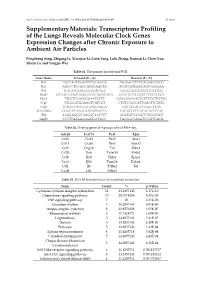
Transcriptome Profiling of the Lungs Reveals Molecular Clock Genes Expression Changes After Chronic Exposure to Ambient Air Particles
Int. J. Environ. Res. Public Health 2017, 14, 0090; doi:10.3390/ijerph14010090 S1 of S6 Supplementary Materials: Transcriptome Profiling of the Lungs Reveals Molecular Clock Genes Expression Changes after Chronic Exposure to Ambient Air Particles Pengcheng Song, Zhigang Li, Xiaoqian Li, Lixin Yang, Lulu Zhang, Nannan Li, Chen Guo, Shuyu Lu and Yongjie Wei Table S1. The primers for real-time PCR. Gene Name Forward (5'—3') Reverse (5'—3') Per1 CAGCAGTGGAGTCTGGAGGA TAGGAGCTCTGAGAAGCGGG Per2 AGCCCTGCAGCATGGAAGTA ACGTCATGAGGAGCCAGGAA Per3 TGTGTTCAAGGGTCCACTGC GGTGCTGGCAACTTCTTTCG Bmal1 CCAAGAAAGTATGGACACAGACAAA GCATTCTTGATCCTTCCTTGGT Clock TTGCTCCACGGGAATCCTT GGAGGGAAAGTGCTCTGTTGTAG Cry1 CTGGCGTGGAAGTCATCGT CTGTCCGCCATTGAGTTCTATG Cry2 TGTCCCTTCCTGTGTGGAAGA GCTCCCAGCTTGGCTTGA REV-ERBα GGGCACAAGCAACATTACCA CACGTCCCCACACACCTTAC Dbp AAGGAGCGCAAGGCAACTCT AGATGTCAAGCCTGCGCGGT Gapdh CCCTTAAGAGGGATGCTGCC TACGGCCAAATCCGTTCACA Table S2. Overlap genes of 4 groups data of RNA-Seq. Adcy9 Cxcl14 Per1 A2m Arntl Cxcl2 Per2 Alas1 Ccl11 Cxcl6 Per3 Alox15 Ccl2 Gng10 Tnf Ddit4 Ccl20 Ifnk Tnfsf10 Efnb2 Ccl9 Il10 Hif3a Epas1 Cry1 Il1b Trim16 Fabp4 Ctf1 Il6 Prl8a2 Tef Cxcl1 Lifr Prl4a1 Table S3. DAVID bioinformatics for functional annotation. Term Count % p-Value Cytokine-cytokine receptor interaction 12 34.2857143 1.27E-10 Chemokine signaling pathway 10 28.5714286 8.32E-09 TNF signaling pathway 7 20 2.31E-06 Circadian rhythm 5 14.2857143 4.03E-06 Herpes simplex infection 8 22.8571429 1.05E-05 Rheumatoid arthritis 6 17.1428571 1.60E-05 Legionellosis 5 14.2857143 5.41E-05 Malaria 5 14.2857143 6.20E-05 Pertussis 5 14.2857143 1.43E-04 African trypanosomiasis 4 11.4285714 3.62E-04 Circadian entrainment 5 14.2857143 4.28E-04 Chagas disease (American 5 14.2857143 6.21E-04 trypanosomiasis) NOD-like receptor signaling pathway 4 11.4285714 0.001137577 Jak-STAT signaling pathway 5 14.2857143 0.001482411 Inflammatory bowel disease (IBD) 4 11.4285714 0.001752567 Int. -

SCIENCE CHINA Spectrin: Structure, Function and Disease
SCIENCE CHINA Life Sciences • REVIEW • December 2013 Vol.56 No.12: 1076–1085 doi: 10.1007/s11427-013-4575-0 Spectrin: Structure, function and disease ZHANG Rui1, ZHANG ChenYu1, ZHAO Qi2 & LI DongHai1* 1Jiangsu Engineering Research Center for microRNA Biology and Biotechnology, State Key Laboratory of Pharmaceutical Biotechnology, School of Life Sciences, Nanjing University, Nanjing 210093, China; 2Institute of Biomedicine and Biotechnology, Shenzhen Institutes of Advanced Technology, Chinese Academy of Sciences, Shenzhen 518055, China Received November 21, 2012; accepted March 20, 2013 Spectrin is a large, cytoskeletal, and heterodimeric protein composed of modular structure of and subunits, it typically contains 106 contiguous amino acid sequence motifs called “spectrin repeats”. Spectrin is crucial for maintaining the stability and structure of the cell membrane and the shape of a cell. Moreover, it contributes to diverse cell functions such as cell adhe- sion, cell spreading, and the cell cycle. Mutations of spectrin lead to various human diseases such as hereditary hemolytic anemia, type 5 spinocerebellar ataxia, cancer, as well as others. This review focuses on recent advances in determining the structure and function of spectrin as well as its role in disease. erythrocyte, spectrin, cell cycle, mass spectrometry, disease Citation: Zhang R, Zhang C Y, Zhao Q, et al. Spectrin: Structure, function and disease. Sci China Life Sci, 2013, 56: 1076–1085, doi: 10.1007/s11427-013-4575-0 Spectrin is a cytoskeletal protein that was first discovered in subunits are encoded by SPTA1 and SPTAN1, respectively. erythrocytes and is important for maintaining the stability, SPTA1 is expressed in erythroid cells. In contrast to SPTA1, structure, and shape of the cell membrane. -

The Trans KCNMA1-M744T and Cis ANXA11-I457V and DYDC2-P123R Variants Are Associated with Familial Dilated Cardiomyopathy
Journal of Molecular and Genetic Purevjav et al., J Mol Genet Med 2020, 14:1 Medicine ISSN: 1747-0862 Research Article Open Access The trans KCNMA1-M744T and cis ANXA11-I457V and DYDC2-P123R Variants are Associated with Familial Dilated Cardiomyopathy Purevjav E1,2*, Zhang W3, Mendsaikhan U3, Gong N3, Martherus R3, Sadek B3, Munkhsaikhan U1,2, Xu F4, Lu L4 and Towbin JA1,2,3,5 1Department of Pediatrics, University of Tennessee Health Science Center, Memphis, TN, United States 2Children’s Foundation Research Institute, Le Bonheur Children’s Hospital, Memphis, TN, United States 3The Heart Institute, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH, United States 4Department of Genetics, Genomics and Informatics, University of Tennessee Health Science Center, Memphis, TN, United States 5Pediatric Cardiology, St. Jude Children's Research Hospital, Memphis, TN, United States *Corresponding author: Dr. EnkhsaikhanPurevjav, 71 S Manassas Street, Room 430K, Memphis, Tennessee 38103, United States, Tel: 901-448-1117, Fax: 901-287- 7460; E-mail: [email protected] Received: March 19, 2020; Accepted: March 23, 2020; Published: March 30, 2020 Copyright: © 2020 Purevjav E, et al. This is an open-access article distributed under the terms of the creative commons attribution license, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. Abstract Objectives: Cardiomyopathies are diseases of heart muscle caused by mutations in cytoskeletal genes. Disease severity and penetrance vary greatly among patients carrying the same mutation(s) and single-gene variants often do not reliably predict cardiomyopathy phenotypes. Background: The chromosome 10q21-q23 locus was previously associated to familial dilated cardiomyopathy (DCM), arrhythmias, heart failure, Wolff-Parkinson-White (WPW) syndrome, mitral valve prolapse (MVP) and/or mitral regurgitation (MR). -

Clinical, Molecular, and Immune Analysis of Dabrafenib-Trametinib
Supplementary Online Content Chen G, McQuade JL, Panka DJ, et al. Clinical, molecular and immune analysis of dabrafenib-trametinib combination treatment for metastatic melanoma that progressed during BRAF inhibitor monotherapy: a phase 2 clinical trial. JAMA Oncology. Published online April 28, 2016. doi:10.1001/jamaoncol.2016.0509. eMethods. eReferences. eTable 1. Clinical efficacy eTable 2. Adverse events eTable 3. Correlation of baseline patient characteristics with treatment outcomes eTable 4. Patient responses and baseline IHC results eFigure 1. Kaplan-Meier analysis of overall survival eFigure 2. Correlation between IHC and RNAseq results eFigure 3. pPRAS40 expression and PFS eFigure 4. Baseline and treatment-induced changes in immune infiltrates eFigure 5. PD-L1 expression eTable 5. Nonsynonymous mutations detected by WES in baseline tumors This supplementary material has been provided by the authors to give readers additional information about their work. © 2016 American Medical Association. All rights reserved. Downloaded From: https://jamanetwork.com/ on 09/30/2021 eMethods Whole exome sequencing Whole exome capture libraries for both tumor and normal samples were constructed using 100ng genomic DNA input and following the protocol as described by Fisher et al.,3 with the following adapter modification: Illumina paired end adapters were replaced with palindromic forked adapters with unique 8 base index sequences embedded within the adapter. In-solution hybrid selection was performed using the Illumina Rapid Capture Exome enrichment kit with 38Mb target territory (29Mb baited). The targeted region includes 98.3% of the intervals in the Refseq exome database. Dual-indexed libraries were pooled into groups of up to 96 samples prior to hybridization. -

270 Genes Genetic Insights Panel
OVER 270 GENES GENETIC INSIGHTS PANEL Dravet Syndrome SCN1A EPILEPSY, Early Infantile epileptic encephalopathy 7 KCNQ2 Early infantile epileptic encephalopathy 11 / Benign familial infantile seizures 3 SCN2A SEIZURES, & OTHER Early infantile epileptic encephalopathy 13 / Benign familial infantile seizures 5 SCN8A NEUROMUSCULAR Ethylmalonic Encephalopathy ETHE1 Familial Infantile Convulsions with Paroxysmal Choreoathetosis PRRT2 CONDITIONS Pyridoxine-Dependent Epilepsy ALDH7A1 Pyridoxal 5'-Phosphate-Dependent Epilepsy PNPO Tuberous Sclerosis Complex (TSC) TSC1, TSC2 Clouston syndrome GJB6 Deafness, Autosomal Dominant, 12 TECTA Deafness, Autosomal Recessive, 6 (DFNB6) TMIE Deafness, Autosomal Recessive, 8 (DFNB8) TMPRSS3 Deafness, Autosomal Recessive, 28 TRIOBP Deafness, Autosomal Recessive, 31 (DFNB31) WHRN Deafness, Autosomal Recessive, 79 TPRN GJB2-Related Hearing Loss GJB2 Hermansky-Pudlak syndrome HPS4 Hermansky-Pudlak Syndrome, Type 1 HPS1 Jervell and Lange-Nielson Syndrome KCNE1 Nonsyndromic Hearing Loss CDH23 HEARING & Ocular Albinism Type I GPR143 Oculocutaneous Albinism Type IV SLC45A2 VISION LOSS Optic Atrophy Type 1 OPA1 Ornithine Aminotransferase Deficiency OAT Pendred Syndrome SLC26A4 Sensorineural Hearing Loss MYO15A Shah-Waardenburg Syndrome SOX10 Usher syndrome type 1C USH1C Usher Syndrome, Type ID CDH23 Usher syndrome type 1G USH1G Usher Syndrome, Type IIA USH2A Usher syndrome type IID WHRN Waardenburg Syndrome PAX3 Alagille Syndrome 1 / Tetralogy of Fallot JAG1 Arrhythmogenic Right Ventricular Cardiomyopathy DSC2 Arrhythmogenic -

A Novel SPTB Gene Mutation in Neonatal Hereditary Spherocytosis: a Case Report and Literature Review
A novel SPTB gene mutation in neonatal hereditary spherocytosis: a case report and literature review Yang Liu Tianjin Children's Hospital Li Song Tianjin Children's Hospital Jianbo Shu Tianjin Children's Hospital Yulian Fang Tianjin Children's Hospital Chao Sun Tianjin Children's Hospital Na Li Tianjin Children's Hospital Geli Liu ( [email protected] ) Tianjin Medical University General Hospital Research article Keywords: Neonate; hereditary spherocytosis; spectrin beta gene; novel mutation Posted Date: August 28th, 2019 DOI: https://doi.org/10.21203/rs.2.10866/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/14 Abstract Background: To enhance our understanding on the diagnosis and treatment of neonatal hereditary spherocytosis (HS). Methods: We summarized the clinical data and gene test results of a neonatal HS caused by a new mutation of SPTB gene. Meanwhile, a comprehensive literature review was performed. Gene sequencing and analysis was carried out for the crucial splicing signals on the exons and introns of the 302 known pathogenic genes (e.g. ANK1, SPTA1, EPB42, SLC4A1 and SPTB) associated with the genetic deciency of erythrocyte. Results: A 26-day-old girl presented jaundice, anemia, an increased count in peripheral blood reticulocyte and spherocyte, as well as positive ndings in the acidied glycerol hemolysis test. Gene sequencing revealed a new mutation of c.3737delA P. (Lys1246fs) in the exon 16 of SPTB (14q23 | NM_000347.5) gene in the patient and her father. The mutation was a frame-shifting mutation, which may result in truncation of beta-haemoglobin in erythrocyte membrane and loss of its normal function, leading to the occurrence of diseases. -

Novel Protein Pathways in Development and Progression of Pulmonary Sarcoidosis Maneesh Bhargava1*, K
www.nature.com/scientificreports OPEN Novel protein pathways in development and progression of pulmonary sarcoidosis Maneesh Bhargava1*, K. J. Viken1, B. Barkes2, T. J. Grifn3, M. Gillespie2, P. D. Jagtap3, R. Sajulga3, E. J. Peterson4, H. E. Dincer1, L. Li2, C. I. Restrepo2, B. P. O’Connor5, T. E. Fingerlin5, D. M. Perlman1 & L. A. Maier2 Pulmonary involvement occurs in up to 95% of sarcoidosis cases. In this pilot study, we examine lung compartment-specifc protein expression to identify pathways linked to development and progression of pulmonary sarcoidosis. We characterized bronchoalveolar lavage (BAL) cells and fuid (BALF) proteins in recently diagnosed sarcoidosis cases. We identifed 4,306 proteins in BAL cells, of which 272 proteins were diferentially expressed in sarcoidosis compared to controls. These proteins map to novel pathways such as integrin-linked kinase and IL-8 signaling and previously implicated pathways in sarcoidosis, including phagosome maturation, clathrin-mediated endocytic signaling and redox balance. In the BALF, the diferentially expressed proteins map to several pathways identifed in the BAL cells. The diferentially expressed BALF proteins also map to aryl hydrocarbon signaling, communication between innate and adaptive immune response, integrin, PTEN and phospholipase C signaling, serotonin and tryptophan metabolism, autophagy, and B cell receptor signaling. Additional pathways that were diferent between progressive and non-progressive sarcoidosis in the BALF included CD28 signaling and PFKFB4 signaling. Our studies demonstrate the power of contemporary proteomics to reveal novel mechanisms operational in sarcoidosis. Application of our workfows in well-phenotyped large cohorts maybe benefcial to identify biomarkers for diagnosis and prognosis and therapeutically tenable molecular mechanisms. -

Novel Mutation in SPTA1 Gene Associated with Severe Hemolytic Anemia
Journal of Pediatrics and Neonatal Care Case Report Open Access Novel mutation in SPTA1 gene associated with severe hemolytic anemia Abstract Volume 8 Issue 5 - 2018 Hereditary spherocytosis (HS), elliptocytosis (HE), and pyropoikilocytosis (HPP) James Polega,1 Jennifer Stumph,2 Archana are caused by mutations in the genes which encode erythrocyte cytoskeletal proteins. 3 4 We report a patient with severe hemolytic anemia with a complex set of mutations, Agarwal, Chi L Braunreiter 1Department of Pediatrics, Spectrum Health Graduate Medical including a novel mutation predicted to cause abnormal splicing of SPTA1 gene, Education, USA highlighting the utility of molecular diagnostics in patients with no identifiable family 2Department of Pathology, Spectrum Health Hospitals, USA history of erythrocyte cytoskeletal disorders. 3Department of Pathology/ARUP Laboratories, University of Utah, USA spectrin, anemia, spherocytosis, elliptocytosis, hereditary Keywords: 4Division of Pediatrics Hematologyand Oncology, Helen De Vos Children’s Hospital, USA Correspondence: Chi L Braunreiter, Division of Pediatrics Hematology and Oncology, Helen De Vos Children’s Hospital, 100 Michigan Street NE, Grand Rapids, Michigan, USA, Tel (616)3912086, Fax (616)3919430, Email [email protected] Received: January 29, 2018 | Published: February 29, 2018 Introduction Osmotic fragility (OF) testing, prior to any packed red blood cell (PRBC) transfusions, was abnormal with increased red blood cell Hereditary spherocytosis (HS), hereditary elliptocytosis (HE), lysis suggesting the presence of spherocytes. However, the eosin-5- and hereditary pyropoikilocytosis (HPP) are caused by mutations in maleimide (EMA) binding test showed a normal staining pattern. Red five genes ANK1, SLC4A1, SPTA1, SPTB and EPB4 which encode blood cell enzyme levels, evaluated prior to any PRBC transfusions, for the erythrocyte cytoskeletal proteins, ankyrin, band 3, α-spectrin, of glucose-6-phosphate dehydrogenase, pyruvate kinase, glucose 1 β-spectrin and protein 4.2, respectively. -
Aagab S00002 Aars S00003 Aars2 S00004 Aass S02483
Test name Code Test name Code Test name Code Test name Code Test name Code Test name Code A ADAR S00053 ALPL S00105 ARSB S00153 BCL10 S02266 C5AR2 S00263 AAGAB S00002 ADCK3 S00054 ALS2 S00106 ARSE * S00154 BCL11A S02167 C5ORF42 S00264 AARS S00003 ADCK4 S00055 ALX3 S00107 ARX S00155 BCL11B S02358 C6 S00265 AARS2 S00004 ADCY10 S02094 ALX4 S00108 ASAH1 S00156 BCOR S00212 C7 S00266 AASS S02483 ADCY3 S02184 AMACR S00109 ASL S00157 BCS1L S00213 C8A S00267 ABAT S02191 ADCY5 S02226 AMELX S02289 ASNS * S02508 BDNF S02509 C8B S00268 ABCA1 S00005 ADGRG1 S00057 AMER1 S00110 ASPA S00158 BDP1 * S00214 C8G S00269 ABCA12 S00006 ADGRG6 S02548 AMH S00111 ASPH S02425 BEAN1 S00215 C8ORF37 S00270 ABCA3 S00007 ADGRV1 S00058 AMHR2 S00112 ASPM S00159 BEST1 S00216 C9 S00271 ABCA4 S00008 ADIPOQ S00059 AMN S00113 ASS1 S00160 BFSP1 S02280 CA2 S00272 ABCA7 S02106 ADIPOR1 * S00060 AMPD1 S02670 ATAD3A * S02196 BFSP2 S00217 CA4 S02303 ABCB11 S00009 ADIPOR2 S00061 AMPD2 S02128 ATCAY S00162 BGN S02633 CA8 S00273 ABCB4 S00010 ADK S02595 AMT S00114 ATF6 S00163 BHLHA9 S00218 CABP2 S00274 ABCB6 S00011 ADNP S02320 ANG S00115 ATIC S02458 BICD2 S00220 CABP4 S00275 ABCB7 S00012 ADSL S00062 ANK1 S00116 ATL1 S00164 BIN1 S00221 CACNA1A S00276 ABCC2 S00013 AFF2 S00063 ANK2 S00117 ATL3 S00165 BLK S00222 CACNA1C * S00277 ABCC6 * S00014 AFG3L2 * S00064 ANKH S00118 ATM S00166 BLM S00223 CACNA1D S00278 ABCC8 S00015 AGA S00065 ANKRD11 * S02140 ATOH7 S02390 BLNK S02281 CACNA1F S00279 ABCC9 S00016 AGBL5 S02452 ANKS6 S00121 ATP13A2 S00168 BLOC1S3 S00224 CACNA1H S00280 ABCD1 * S00017 AGK * -

ADX609 3301 Athena Newborndx Gene Panel Update 3-30-20.Indd
NewbornDx™ Advanced Sequencing Evaluation When time to diagnosis matters, the NewbornDx™ Advanced Sequencing Evaluation from Athena Diagnostics delivers rapid, 3- to 7-day results on a targeted 1,722-genes. A2ML1 ALAD ATM CAV1 CLDN19 CTNS DOCK7 ETFB FOXC2 GLUL HOXC13 JAK3 AAAS ALAS2 ATP1A2 CBL CLIC2 CTRC DOCK8 ETFDH FOXE1 GLYCTK HOXD13 JUP AARS2 ALDH18A1 ATP1A3 CBS CLMP CTSA DOK7 ETHE1 FOXE3 GM2A HPD KANK1 AASS ALDH1A2 ATP2B3 CC2D2A CLN3 CTSD DOLK EVC FOXF1 GMPPA HPGD K ANSL1 ABAT ALDH3A2 ATP5A1 CCDC103 CLN5 CTSK DPAGT1 EVC2 FOXG1 GMPPB HPRT1 KAT6B ABCA12 ALDH4A1 ATP5E CCDC114 CLN6 CUBN DPM1 EXOC4 FOXH1 GNA11 HPSE2 KCNA2 ABCA3 ALDH5A1 ATP6AP2 CCDC151 CLN8 CUL4B DPM2 EXOSC3 FOXI1 GNAI3 HRAS KCNB1 ABCA4 ALDH7A1 ATP6V0A2 CCDC22 CLP1 CUL7 DPM3 EXPH5 FOXL2 GNAO1 HSD17B10 KCND2 ABCB11 ALDOA ATP6V1B1 CCDC39 CLPB CXCR4 DPP6 EYA1 FOXP1 GNAS HSD17B4 KCNE1 ABCB4 ALDOB ATP7A CCDC40 CLPP CYB5R3 DPYD EZH2 FOXP2 GNE HSD3B2 KCNE2 ABCB6 ALG1 ATP8A2 CCDC65 CNNM2 CYC1 DPYS F10 FOXP3 GNMT HSD3B7 KCNH2 ABCB7 ALG11 ATP8B1 CCDC78 CNTN1 CYP11B1 DRC1 F11 FOXRED1 GNPAT HSPD1 KCNH5 ABCC2 ALG12 ATPAF2 CCDC8 CNTNAP1 CYP11B2 DSC2 F13A1 FRAS1 GNPTAB HSPG2 KCNJ10 ABCC8 ALG13 ATR CCDC88C CNTNAP2 CYP17A1 DSG1 F13B FREM1 GNPTG HUWE1 KCNJ11 ABCC9 ALG14 ATRX CCND2 COA5 CYP1B1 DSP F2 FREM2 GNS HYDIN KCNJ13 ABCD3 ALG2 AUH CCNO COG1 CYP24A1 DST F5 FRMD7 GORAB HYLS1 KCNJ2 ABCD4 ALG3 B3GALNT2 CCS COG4 CYP26C1 DSTYK F7 FTCD GP1BA IBA57 KCNJ5 ABHD5 ALG6 B3GAT3 CCT5 COG5 CYP27A1 DTNA F8 FTO GP1BB ICK KCNJ8 ACAD8 ALG8 B3GLCT CD151 COG6 CYP27B1 DUOX2 F9 FUCA1 GP6 ICOS KCNK3 ACAD9 ALG9