Isotopic Neutron Sources for Neutron Activation Analysis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Production of Radionuclides
CHAPTER 15 Production of Radionuclides Contents 15.1. General considerations 389 15.2. Irradiation yields 390 15.3. Second-order reactions 393 15.4. Target considerations 397 15.4.1. Physical properties 397 15.4.2. Chemical properties 398 15.5. Product specifications 399 15.5.1. Radiochemical processing 399 15.5.2. Specific activity 400 15.5.3. Labeling 400 15.5.4. Radiochemical purity 401 15.6. Recoil separations 402 15.6.1. Target recoil products 402 15.6.2. Hot atom reactions 403 15.6.3. The Szilard-Chalmers process 404 15.7. Fast radiochemical separations 406 15.7.1. Production of 11C labeled compounds 407 15.7.2. Auto-batch procedures 407 15.7.3. On-line procedures: gas-phase separation 409 15.7.4. On-line procedures: solvent extraction 412 15.7.5. Mass separator procedures 412 15.8. Exercises 412 15.9. Literature 414 This chapter discusses production of radionuclides for beneficial use in science, medicine and technology. The nuclear fundamentals for the production processes have been given in Chapters 11 to 14. The formation of radionuclides is discussed in several Chapters: e.g. cosmogenic reactions leading to the formation of short-lived radionuclides in nature (Ch. 5 and 10); thermonuclear reactions leading to the formation of long-lived radioactivity in the universe (Ch. 17); the synthesis of trans-uranium elements (Ch. 16 and 19-21). The production and isolation of separated fission products is treated separately (Ch. 19-21). This chapter discusses aspects of fundamental importance to the production of radionuclides by a variety of methods. -

MASTER 9700 South Cass Avenue Argonne, Illinois 60439 USA
ANL/KDH—SO DE84 001440 ANL/NDM-80 NEUTRON TOTAL CROSS SECTION MEASUREMENTS IN THE ENERGY REGION FROM 47 key to 20 MeV* by W. P. Poenitz and J. F. Whalon Applied Physics Division May, 1983 *This work supported by the U.S. Department of Energy Argonne National Laboratory MASTER 9700 South Cass Avenue Argonne, Illinois 60439 USA fflSTRtBtfUOU OF miS DOCUMENT IS UNLIMITED NUCLEAR DATA AMD MEASUREMENTS SERIES The Nuclear Data and Measurements Series presents results of studies in the field of microscopic nuclear data. The primary objective is the dissemination of information in the comprehensive form required for nuclear technology applications. This Series is devoted to: a) measured microscopic nuclear parameters, b) experimental techniques and facilities employed in measurements, c) the analysis, correlation and interpretation of nuclear data, and d) the evaluation of nuclear data. Contributions to this Series are reviewed to assure technical competence and, unless otherwise stated, the contents can be formally referenced. This Series does not supplant formal journal publication but it does provide the more extensive informa- tion required for technological applications (e.g., tabulated numerical data) in a timely manner. DISCLAIMER This report was prepared as an account of work sponsored by an agency of the United States Government. Neither the United States Government nor any agency thereof, nor any of their employees, makes any warranty, express or implied, or assumes any legal liability or retpoosi- bility for the accuracy, completeness, or usefulness of any information, apparatus, product, or process disclosed, or represents that its use would not infringe privately owned rights. Refer- ence herein to any specific commercial product, process, or service by trade name, trademark, manufacturer, or otherwise does not necessarily constitute or imply its endorsement, recom- mendation, or favoring by the United States Government or any agency thereof. -

Uses of Isotopic Neutron Sources in Elemental Analysis Applications
EG0600081 3rd Conference on Nuclear & Particle Physics (NUPPAC 01) 20 - 24 Oct., 2001 Cairo, Egypt USES OF ISOTOPIC NEUTRON SOURCES IN ELEMENTAL ANALYSIS APPLICATIONS A. M. Hassan Department of Reactor Physics Reactors Division, Nuclear Research Centre, Atomic Energy Authority. Cairo-Egypt. ABSTRACT The extensive development and applications on the uses of isotopic neutron in the field of elemental analysis of complex samples are largely occurred within the past 30 years. Such sources are used extensively to measure instantaneously, simultaneously and nondestruclively, the major, minor and trace elements in different materials. The low residual activity, bulk sample analysis and high accuracy for short lived elements are improved. Also, the portable isotopic neutron sources, offer a wide range of industrial and field applications. In this talk, a review on the theoretical basis and design considerations of different facilities using several isotopic neutron sources for elemental analysis of different materials is given. INTRODUCTION In principle there are two ways to use neutrons for elemental and isotopic abundance analysis in samples. One is the neutron activation analysis which we call it the "off-line" where the neutron - induced radioactivity is observed after the end of irradiation. The other one we call it the "on-line" where the capture gamma-rays is observed during the neutron bombardment. Actually, the sequence of events occurring during the most common type of nuclear reaction used in this analysis namely the neutron capture or (n, gamma) reaction, is well known for the people working in this field. The neutron interacts with the target nucleus via a non-elastic collision, a compound nucleus forms in an excited state. -

PGNAA Neutron Source Moderation Setup Optimization
Submitted to ‘Chinese Physics C PGNAA neutron source moderation setup optimization Zhang Jinzhao1(张金钊)Tuo Xianguo1(庹先国) (1.Chengdu University of Technology Applied Nuclear Techniques in Geoscience Key Laboratory of Sichuan Province,Chengdu 610059,China) Abstract: Monte Carlo simulations were carried out to design a prompt γ-ray neutron activation analysis (PGNAA) thermal neutron output setup using MCNP5 computer code. In these simulations the moderator materials, reflective materials and structure of the PGNAA 252Cf neutrons of thermal neutron output setup were optimized. Results of the calcuations revealed that the thin layer paraffin and the thick layer of heavy water moderated effect is best for 252Cf neutrons spectrum. The new design compared with the conventional neutron source design, the thermal neutron flux and rate were increased by 3.02 times and 3.27 times. Results indicate that the use of this design should increase the neutron flux of prompt gamma-ray neutron activation analysis significantly. Key word: PGNAA; neutron source; thermal neutron; moderation; reflection 1. Introduction study, Monte Carlo calculation was carried out for the Prompt gamma ray neutron activation analysis design of a 252Cf neutron source moderation setup for the (PGNAA) is a rapid, nondestructive, powerful analysis cement samples[7]. The model of Monte Carlo multielemental analysis technique, large samples of some simulation was verified by experiment[8, 9].We improve minor, trace light elements and is used in industrial the thermal neutron source yield rate of 252Cf neutron by control[1-5]. In a PGNAA analysis, the sample nuclear the PGNAA neutron source structure to the design. The composition is determined from prompt gamma rays calculation results for the new design were compared which produced through neutron inelastic scattering and with the previous, example: themal neutron flux rate, fast thermal neutron capture. -
![Arxiv:1506.05417V2 [Physics.Ins-Det] 28 Jul 2016](https://docslib.b-cdn.net/cover/1390/arxiv-1506-05417v2-physics-ins-det-28-jul-2016-561390.webp)
Arxiv:1506.05417V2 [Physics.Ins-Det] 28 Jul 2016
http://dx.doi.org/10.1016/j.apradiso.2016.06.032 A precise method to determine the activity of a weak neutron source using a germanium detector M. J. M. Dukea, A. L. Hallinb, C. B. Kraussb, P. Mekarskib,∗, L. Sibleyb aSLOWPOKE Nuclear Reactor Facility, University of Alberta, Edmonton, AB T6G 2G7, Canada bDepartment of Physics, University of Alberta, Edmonton, AB T6G 2E1, Canada Abstract A standard high purity germanium (HPGe) detector was used to determine the previously unknown neutron activity of a weak americium-beryllium (AmBe) neutron source. γ rays were created through 27Al(n,n0), 27Al(n,γ) and 1H(n,γ) reactions induced by the neutrons on aluminum and acrylic disks, respectively. These γ rays were measured using the HPGe detector. Given the unorthodox experimental arrangement, a Monte Carlo simulation was developed to model the efficiency of the detector system to determine the neutron activity from the measured γ rays. The activity of our neutron source was determined to be 307.4 ± 5.0 n/s and is consistent for the different neutron-induced γ rays. Keywords: neutron activation, germanium detector, simulation, spectroscopy, activity determination 1. Introduction As neutrons are difficult to detect, determining the absolute activity of a neutron source is challenging. This difficulty increases as the activity of the source decreases. Sophisticated techniques exist for neutron activity measure- ments, including the manganese bath technique[1], proton recoil techniques[2] and the use of 3He proportional counters[3]; nevertheless, the development of a method utilizing commonly available high purity germanium (HPGe) detectors would be advantageous. HPGe's are an industry standard for measuring γ ray energies to high preci- sion. -

Project Note Weston Solutions, Inc
PROJECT NOTE WESTON SOLUTIONS, INC. To: Canadian Radium & Uranium Corp. Site File Date: June 5, 2014 W.O. No.: 20405.012.013.2222.00 From: Denise Breen, Weston Solutions, Inc. Subject: Determination of Significant Lead Concentrations in Sediment Samples References 1. New York State Department of Environmental Conservation. Technical Guidance for Screening Contaminated Sediments. March 1998. [45 pages] 2. U.S. Environmental Protection Agency (EPA) Office of Emergency Response. Establishing an Observed Release – Quick Reference Fact Sheet. Federal Register, Volume 55, No. 241. September 1995. [7 pages] 3. International Union of Pure and Applied Chemistry, Inorganic Chemistry Division Commission on Atomic Weights and Isotopic Abundances. Atomic Weights of Elements: Review 2000. 2003. [120 pages] WESTON personnel collected six sediment samples (including one environmental duplicate sample) from five locations along the surface water pathway of the Canadian Radium & Uranium Corp. (CRU) site in May 2014. The sediment samples were analyzed for Target Analyte List (TAL) Metals and Stable Lead Isotopes. 1. TAL Lead Interpretation: In order to quantify the significance for Lead, Thallium and Mercury the following was performed: 1. WESTON personnel tabulated all available TAL Metal data from the May 2014 Sediment Sampling event. 2. For each analyte of concern (Lead, Thallium, and Mercury), the highest background concentration was selected and then multiplied by three. This is the criteria to find the significance of site attributable release as per Hazard Ranking System guidelines. 3. One analytical lead result (2222-SD04) of 520 mg/kg (J) was qualified with an unknown bias. In accordance with US EPA document “Using Data to Document an Observed Release and Observed Contamination”, 2222-SD03 lead concentration was adjusted by dividing by the factor value for lead of 1.44 to equal 361 mg/kg. -

Lawrence Berkeley National Laboratory Recent Work
Lawrence Berkeley National Laboratory Recent Work Title RADIOCHEMICAL AND SPECTROMETER STUDIES OF SEVERAL NEUTRON-DEFICIENT ZIRCONIUM ISOTOPES AND THEIR DECAY PRODUCTS Permalink https://escholarship.org/uc/item/5ck8q7xw Authors Hyde, Earl K. O'Kelley, Grover D. Publication Date 1950-12-28 eScholarship.org Powered by the California Digital Library University of California UCRL- .Jt)AJ,-.64.. _.; >-w .J w ... ~ ,• 0:: w m I . ;•· <( z 0:: 0 LL ..J u<( LL 0 >- 1-- TWO-WEEK LOAN COPY (/) This is a Library Circulating Copy 0:: which may be borrowed for two weeks. w For a personal retention copy, call > Tech. Info. Diuision, Ext. 5545 z '.-o:-. :J ~~ RADIATION LABORATORY DISCLAIMER This document was prepared as an account of work sponsored by the United States Government. While this document is believed to contain correct information, neither the United States Government nor any agency thereof, nor the Regents of the University of California, nor any of their employees, makes any warranty, express or implied, or assumes any legal responsibility for the accuracy, completeness, or usefulness of any · information, apparatus, product, or process disclosed, or represents that its use would not infringe privately owned rights. Reference herein to any specific commercial product, process, or service by its trade name, trademark, manufacturer, or otherwise, does not necessarily constitute or imply its endorsement, recommendation, or favoring by the United States Government or any agency thereof, or the Regents of the University of California. The views and opinions of authors expressed herein do not necessarily state or reflect those of the United States Government or any agency thereof or the Regents of the University of California. -

Technische Universität München Physik-Department Study of The
Technische Universität München Physik-Department Study of the biokinetics of zirconium isotopes in humans and its relevance to internal dosimetry Matthias Greiter Vollständiger Abdruck der von der Fakultät für Physik der Technischen Universität München zur Erlangung des akademischen Grades eines Doktors der Naturwissenschaften genehmigten Dissertation. Vorsitzender: Univ.-Prof. Dr. J. L. van Hemmen Prüfer der Dissertation: 1. Hon.-Prof. Dr. H.G. Paretzke 2. Univ.-Prof. Dr. F. von Feilitzsch Die Dissertation wurde am 27.09.2007 bei der Technischen Universität München eingereicht und durch die Fakultät für Physik am 22.04.2008 angenommen. i Author contact information: Matthias Greiter Institute of Radiation Protection Helmholtz Zentrum München German Research Center for Environmental Health (GmbH) Ingolstaedter Landstrasse 1 85764 Neuherberg Germany E-mail: [email protected] ii Table of contents Abstract ...................................................................................................................................... 1 List of acronyms, symbols and abbreviations ............................................................................ 2 1 Aim of the study......................................................................................................... 3 1.1 Natural occurrence of zirconium compounds ............................................................ 3 1.2 Applications of stable zirconium................................................................................ 5 1.3 Applications -

Neutron Sources
Neutron Sources Nadia Fomin Fundamental Neutron Physics Summer School Knoxville, TN 2015 Much content “borrowed” from Kevin Anderson, Mike Snow, Geoff 1 Greene, Scott Dewey, Mike Moncko, Jack Carpenter, and many others http://knoxbeerweek.com/ Neutron Sources Nadia Fomin Fundamental Neutron Physics Summer School Knoxville, TN 2015 Much content “borrowed” from Kevin Anderson, Mike Snow, Geoff 3 Greene, Scott Dewey, Mike Moncko, Jack Carpenter, and many others What is neutron physics ? Research which uses “low energy” neutrons from nuclear reactors and accelerator-driven spallation sources to address questions in nuclear, particle, and astrophysics Neutron Properties -22 Electric charge: qn=0, electrically neutral [qn<10 e] -5 -25 2 Size: rn~10 Angstrom=1 Fermi [area~ 10 cm =0.1 “barn”] Internal Structure: quarks [ddu, md~ mu~few MeV ] + gluons Spin: sn= 1/2 [Fermi statistics] Magnetic Dipole Moment: mn/ mp = -0.68497935(17) -26 Electric Dipole Moment: zero[dn < 10 e-cm] Mass: mn=939.566 MeV [mn> mp+ me, neutrons can decay] Lifetime: tn=880ish (depends on whom you ask) Neutrons are hard to get Neutrons are bound in nuclei, need several MeV for liberation E E=0 We want E~kT~25 meV (room temperature) VNN How to slow down a heavy neutral particle with Mn= Mp ? Lots of collisions… p n [1/2]N=(1 MeV)/(25 meV) for N collisions E 0 E/2 Neutrons are unstable when free->they can’t be accumulated easily Neutrons: Fast and Furious. And slow and gentle . Thermal ~25meV (2200m/s, λT=1.8Å) . Cold 50μeV-25meV . Very cold 2x10-7 - 5x10-5 eV . -

Development of a National Neutron Database for Nuclear Technology
- 7 - XA9744992 DEVELOPMENT OF A NATIONAL NEUTRON DATABASE FOR NUCLEAR TECHNOLOGY A. V. Ignatyuk, V.N. Kononov, B.D. Kuzminov, V.N. Manokhin M.N. Nikolaev, B.I. Furzov State Research Centre of the Russian Federation Institute of Physics and Power Engineering, Obninsk Abstract This paper describes the stages of a many years activities at the IPPE consisting of the measurement, theoretical description and evaluation of neutron data, and of the establishment of a national data bank of neutron data for nuclear technology. A list of libraries which are stored at the Nuclear Data Centre is given. Introduction Currently, nuclear technology has found wide applications in a number of fields in science and technology consisting of: - nuclear energy (nuclear fission reactors, nuclear fission reactors, radioisotopic sources) - nuclear transmutation (production of radioisotopes for medical and other applications, burn-up of long-lived radio nuclides, radiation alloying of semiconducting materials, etc..) - nuclear instrumentation in industry (activation analysis, nuclear micro-analysis, control of fissile materials, etc..) - scientific investigations (nuclear physics, nuclear astrophysics, medicine, radiobiology, agro-radiology, ecology, etc.). In the sphere of nuclear technology, a number of processes use more than 500 stable and radioactive nuclides in a broad energy range. To ascertain the proper functioning of nuclear installations which have various functions and applications in industrial projects and scientific investigations, there is a need for nuclear data that characterize the properties of nuclear reactions. The reliability of radiation and nuclear safety, the ecological acceptance, and the economical planning of nuclear installations, all depend on the completeness, quality and accuracy of nuclear data. -
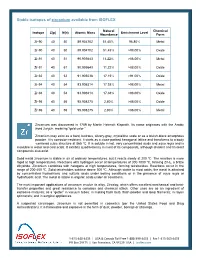
Stable Isotopes of Zirconium Available from ISOFLEX
Stable isotopes of zirconium available from ISOFLEX Natural Chemical Isotope Z(p) N(n) Atomic Mass Enrichment Level Abundance Form Zr-90 40 50 89.904702 51.45% 96.80% Metal Zr-90 40 50 89.904702 51.45% >98.00% Oxide Zr-91 40 51 90.905643 11.22% >88.00% Metal Zr-91 40 51 90.905643 11.22% >88.00% Oxide Zr-92 40 52 91.905038 17.15% >91.00% Oxide Zr-94 40 54 93.906314 17.38% >98.00% Metal Zr-94 40 54 93.906314 17.38% >98.00% Oxide Zr-96 40 56 95.908275 2.80% >86.00% Oxide Zr-96 40 56 95.908275 2.80% >86.00% Metal Zirconium was discovered in 1789 by Martin Heinrich Klaproth. Its name originates with the Arabic word zargun, meaning “gold color.” Zirconium may exist as a hard, lustrous, silvery-gray, crystalline scale or as a bluish-black amorphous powder. It is corrosion-resistant. It starts as a close-packed hexagonal lattice and transforms to a body -centered cubic structure at 865 ºC. It is soluble in hot, very concentrated acids and aqua regia and is insoluble in water and cold acids. It exhibits quadrivalency in most of its compounds, although divalent and trivalent compounds also exist. Solid metal zirconium is stable in air at ordinary temperatures, but it reacts slowly at 200 ºC. The reaction is more rapid at high temperatures. Reactions with hydrogen occur at temperatures of 300-1000 ºC, forming ZrH2, a brittle dihydride. Zirconium combines with halogens at high temperatures, forming tetrahalides. -

Chapter 3 Gamma-Ray and Neutron Sources R.J. Holmes
123 CHAPTER 3 GAMMA-RAY AND NEUTRON SOURCES R.J. HOLMES 125 1. GAMMA-RAY SOURCES Most y-ray sources in commercial use do not occur in nature because their half-lives are small compared with geological times. They must be produced from naturally occurring nuclides by a suitable nucle?r reaction; often this is by irradiation in a nuclear reactor. Some examples of radioisotope production are given below: 59Co (n,y)60Co T^ 5.26 y 123Sb (n,y)mSb T^ 60 d 6Li (n,a)3H T^ 12.3 y 55Mn (p,n)55Fe T, 2.7 y Commercially available sources are sealed in chemically inert capsules. The choice of the most suitable source for a particular application usually depends on the energy of the y-rays that are emitted and on the half-life of the radioisotope. In many applications, a monoenergetic source of long half-life is preferred. Calibration corrections for source decay can be made using the familiar equation - 0.693t/T, = Io e where I is the initial source intensity/ I(t) is its intensity at time o t, and T, is the half-life. Selection of the appropriate y-ray energy depends on such criteria as the energy threshold for a desired nuclear reaction and whether absorption should be due predominantly to the photoelectric effect or Compton scattering. Table 1 lists the commonly used y-ray sources together with their y-ray energies and half-lives. 2. X-RAY SOURCES Sources of radiation below an energy of about 150 keV are usually referred to as X-ray sources, although technically some of them are low energy y-ray sources, e.g.