Corrupted DNA-Binding Specificity and Ectopic Transcription Underpin Dominant Neomorphic Mutations in KLF/SP Transcription Factors Melissa D
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Mediator of DNA Damage Checkpoint 1 (MDC1) Is a Novel Estrogen Receptor Co-Regulator in Invasive 6 Lobular Carcinoma of the Breast 7 8 Evelyn K
bioRxiv preprint doi: https://doi.org/10.1101/2020.12.16.423142; this version posted December 16, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. 1 Running Title: MDC1 co-regulates ER in ILC 2 3 Research article 4 5 Mediator of DNA damage checkpoint 1 (MDC1) is a novel estrogen receptor co-regulator in invasive 6 lobular carcinoma of the breast 7 8 Evelyn K. Bordeaux1+, Joseph L. Sottnik1+, Sanjana Mehrotra1, Sarah E. Ferrara2, Andrew E. Goodspeed2,3, James 9 C. Costello2,3, Matthew J. Sikora1 10 11 +EKB and JLS contributed equally to this project. 12 13 Affiliations 14 1Dept. of Pathology, University of Colorado Anschutz Medical Campus 15 2Biostatistics and Bioinformatics Shared Resource, University of Colorado Comprehensive Cancer Center 16 3Dept. of Pharmacology, University of Colorado Anschutz Medical Campus 17 18 Corresponding author 19 Matthew J. Sikora, PhD.; Mail Stop 8104, Research Complex 1 South, Room 5117, 12801 E. 17th Ave.; Aurora, 20 CO 80045. Tel: (303)724-4301; Fax: (303)724-3712; email: [email protected]. Twitter: 21 @mjsikora 22 23 Authors' contributions 24 MJS conceived of the project. MJS, EKB, and JLS designed and performed experiments. JLS developed models 25 for the project. EKB, JLS, SM, and AEG contributed to data analysis and interpretation. SEF, AEG, and JCC 26 developed and performed informatics analyses. MJS wrote the draft manuscript; all authors read and revised the 27 manuscript and have read and approved of this version of the manuscript. -
Drosophila and Human Transcriptomic Data Mining Provides Evidence for Therapeutic
Drosophila and human transcriptomic data mining provides evidence for therapeutic mechanism of pentylenetetrazole in Down syndrome Author Abhay Sharma Institute of Genomics and Integrative Biology Council of Scientific and Industrial Research Delhi University Campus, Mall Road Delhi 110007, India Tel: +91-11-27666156, Fax: +91-11-27662407 Email: [email protected] Nature Precedings : hdl:10101/npre.2010.4330.1 Posted 5 Apr 2010 Running head: Pentylenetetrazole mechanism in Down syndrome 1 Abstract Pentylenetetrazole (PTZ) has recently been found to ameliorate cognitive impairment in rodent models of Down syndrome (DS). The mechanism underlying PTZ’s therapeutic effect is however not clear. Microarray profiling has previously reported differential expression of genes in DS. No mammalian transcriptomic data on PTZ treatment however exists. Nevertheless, a Drosophila model inspired by rodent models of PTZ induced kindling plasticity has recently been described. Microarray profiling has shown PTZ’s downregulatory effect on gene expression in fly heads. In a comparative transcriptomics approach, I have analyzed the available microarray data in order to identify potential mechanism of PTZ action in DS. I find that transcriptomic correlates of chronic PTZ in Drosophila and DS counteract each other. A significant enrichment is observed between PTZ downregulated and DS upregulated genes, and a significant depletion between PTZ downregulated and DS dowwnregulated genes. Further, the common genes in PTZ Nature Precedings : hdl:10101/npre.2010.4330.1 Posted 5 Apr 2010 downregulated and DS upregulated sets show enrichment for MAP kinase pathway. My analysis suggests that downregulation of MAP kinase pathway may mediate therapeutic effect of PTZ in DS. Existing evidence implicating MAP kinase pathway in DS supports this observation. -

A Core Erythroid Transcriptional Network Is Repressed by a Master Regulator of Myelo-Lymphoid Differentiation
A core erythroid transcriptional network is repressed by a master regulator of myelo-lymphoid differentiation Sandeep N. Wontakala,1, Xingyi Guob,1, Cameron Smithc,2, Thomas MacCarthyc,d,2, Emery H. Bresnicke, Aviv Bergmanc, Michael P. Snyderf,g, Sherman M. Weissmanh,3, Deyou Zhengb,i,3, and Arthur I. Skoultchia,3 Departments of aCell Biology, bNeurology, cSystems and Computational Biology, and iGenetics and Neuroscience, Albert Einstein College of Medicine, Bronx, NY 10461; dDepartment of Applied Mathematics and Statistics, State University of New York, Stony Brook, NY 11794; eWisconsin Institutes for Medical Research, University of Wisconsin School of Medicine and Public Health, Madison, WI 53705; hDepartment of Genetics, Yale University School of Medicine, New Haven, CT 06520; fDepartment of Molecular, Cellular, and Developmental Biology, Yale University, New Haven, CT 06520; and gDepartment of Genetics, Stanford University, Stanford, CA 94305 Contributed by Sherman M. Weissman, December 29, 2011 (sent for review October 10, 2011) Two mechanisms that play important roles in cell fate decisions are PU.1, is a negative regulator of terminal erythroid differentiation control of a “core transcriptional network” and repression of alter- (16–19). Surprisingly, PU.1 was found to occupy more genes in native transcriptional programs by antagonizing transcription fac- erythroid progenitors than the three erythroid-promoting factors tors. Whether these two mechanisms operate together is not (20). However, the extent of overlap between the genes bound by known. Here we report that GATA-1, SCL, and Klf1 form an erythroid PU.1 and the three erythroid factors is not known. core transcriptional network by co-occupying >300 genes. -

Roles of Krüppel Like Factors Klf1, Klf2, and Klf4 in Embryonic Beta-Globin Gene Expression
Virginia Commonwealth University VCU Scholars Compass Theses and Dissertations Graduate School 2009 ROLES OF KRÜPPEL LIKE FACTORS KLF1, KLF2, AND KLF4 IN EMBRYONIC BETA-GLOBIN GENE EXPRESSION Yousef Alhashem Virginia Commonwealth University Follow this and additional works at: https://scholarscompass.vcu.edu/etd Part of the Medical Genetics Commons © The Author Downloaded from https://scholarscompass.vcu.edu/etd/1880 This Thesis is brought to you for free and open access by the Graduate School at VCU Scholars Compass. It has been accepted for inclusion in Theses and Dissertations by an authorized administrator of VCU Scholars Compass. For more information, please contact [email protected]. Virginia Commonwealth University School of Medicine This is to certify that the thesis prepared by Yousef N. Alhashem entitled “Roles Of Krüppel Like Factors KLF1, KLF2, And KLF4 In Embryonic Beta-Globin Gene Expression” has been approved by the student advisory committee as a satisfactory for the completion of the thesis reqirement for the degree of Master of Science. _____________________________________________ Joyce A. Lloyd, Ph.D., Director of Thesis, School of Medicine _____________________________________________ Rita Shiang, Ph.D., School of Medicine _____________________________________________ David C. Williams Jr., Ph.D., School of Medicine _____________________________________________ Paul B. Fisher, Ph.D., Chair, Department of Human and Molecular Genetics _____________________________________________ Jerome F. Strauss, III, M.D., Ph.D., Dean, School of Medicine _____________________________________________ F. Douglas Boudinot, Ph.D., Dean, Graduate School _____________________________________________ Date © Yousef Nassir Alhashem 2009 ROLES OF KRÜPPEL LIKE FACTORS KLF1, KLF2, AND KLF4 IN EMBRYONIC BETA-GLOBIN GENE EXPRESSION A thesis submitted in partial fulfillment of the requirements for the degree of Master of Science at Virginia Commonwealth University By Yousef Nassir Alhashem, B.Sc. -

Analysis of Long Range Gene Regulation in Drosophila; Insights
Analysis Of Long Range Gene Regulation In Drosophila; Insights From The dPax2 sparking Enhancer by Nicole C. Evans A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy (Cell and Developmental Biology) in the University of Michigan 2012 Doctoral Committee: Associate Professor Scott Barolo, Chair Professor Kenneth Cadigan Assistant Professor Catherine Collins Assistant Professor Kentaro Nabeshima DEDICATION To my family, who have been unfailingly supportive through my entire education and the doctoral experience. My Grandpa Vern, who assigned my sister and me our future careers at age ten, but would have loved me just the same if I had not followed his instructions. To my Grandma Judy, who can make anything positive and has always been there for me with her love and encouragement. To my sister Katelyn, whose sense of humor and sassiness makes my world bright. Especially to my parents, my Dad who taught me to approach every problem with a creative and open mind and my Mom who inspired me to learn and teach others. Without their laughter, love, and support I would not be here chasing my dreams. ii ACKNOWLEDGMENTS I deeply thank Christina Swanson for her friendship and enabling me to pursue a study of the sparking enhancer. I am grateful to Amy Strom and Alex Chappell whose smiling faces and eager brains remind me why I love science every day, for their contributions to this work, and for all the mini preps. iii TABLE OF CONTENTS DEDICATION………..…………………………………………………………..………ii ACKNOWLEDGEMENTS………………………….…………………..……………...iii LIST O F FIGURES…………..……………………………………………………….xiii LIST OF TABLES……………………………………………………………………..xvii CHAPTER 1 SPARKLING INSIGHTS INTO ENHANCER, STRUCTURE, FUNCTION, AND EVOLUTION ........................................................................... -

Genetic Disarray Follows Mutant KLF1-E325K Expression in A
ARTICLE Red Cell Biology & its Disorders Genetic disarray follows mutant KLF1-E325K Ferrata Storti Foundation expression in a congenital dyserythropoietic anemia patient Lilian Varricchio,1 Antanas Planutis,2 Deepa Manwani,3 Julie Jaffray,4 W. Beau Mitchell,5 Anna Rita Migliaccio1,6,* and James J. Bieker1,2,7,8,* 1Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, NY, USA; 2Department of Cell, Developmental, and Regenerative Biology, Icahn School of Haematologica 2019 Medicine at Mount Sinai, New York, NY, USA; 3Division of Hematology/Oncology, The Volume 104(12):2372-2381 Children's Hospital at Montefiore, Albert Einstein College of Medicine, Bronx, NY, USA; 4Children's Hospital Los Angeles, University of Southern California Keck School of Medicine, Los Angeles, CA, USA; 5Department of Pediatrics, Icahn School of Medicine at Mount Sinai, New York, NY, USA; 6Dipartimento di Scienze Biomediche e NeuroMotorie, Alma Mater Studiorum, Università di Bologna, Bologna, Italy; 7Black Family Stem Cell Institute, Icahn School of Medicine at Mount Sinai, New York, NY, USA and 8Mindich Child Health and Development Institute, Icahn School of Medicine at Mount Sinai, New York, NY, USA *ARM and JJB contributed equally as senior authors on this work. ABSTRACT ongenital dyserythropoietic anemia type IV is caused by a heterozy- gous mutation, Glu325Lys (E325K), in the KLF1 transcription factor. CMolecular characteristics of this disease have not been clarified, part- ly due to its rarity. We expanded erythroid cells from a patient’s peripheral blood and analyzed its global expression pattern. We find that a large num- ber of erythroid pathways are disrupted, particularly those related to mem- brane transport, globin regulation, and iron utilization. -

Multiple Functions of Autophagy in Vascular Calcification
Zhou et al. Cell Biosci (2021) 11:159 https://doi.org/10.1186/s13578-021-00639-9 Cell & Bioscience REVIEW Open Access Multiple functions of autophagy in vascular calcifcation Xin Zhou1†, Sui‑Ning Xu2†, Shu‑Tong Yuan1†, Xinjuan Lei3, Xiaoying Sun4, Lu Xing1, Hui‑Jin Li1, Chun‑Xia He1, Wei Qin1, Dong Zhao1, Peng‑Quan Li1, Edward Moharomd5*, Xuehong Xu3* and Hui‑Ling Cao1* Abstract Background: Vascular calcifcation is a closely linked to cardiovascular diseases, such as atherosclerosis, chronic kidney disease, diabetes, hypertension and aging. The extent of vascular calcifcation is closely correlate with adverse clinical events and cardiovascular all‑cause mortality. The role of autophagy in vascular calcifcation is complex with many mechanistic unknowns. Methods: In this review, we analyze the current known mechanisms of autophagy in vascular calcifcation and dis‑ cuss the theoretical advantages of targeting autophagy as an intervention against vascular calcifcation. Results: Here we summarize the functional link between vascular calcifcation and autophagy in both animal models of and human cardiovascular disease. Firstly, autophagy can reduce calcifcation by inhibiting the osteogenic dif‑ ferentiation of VSMCs related to ANCR, ERα, β‑catenin, HIF‑1a/PDK4, p62, miR‑30b, BECN1, mTOR, SOX9, GHSR/ERK, and AMPK signaling. Conversely, autophagy can induce osteoblast diferentiation and calcifcation as mediated by CREB, degradation of elastin, and lncRNA H19 and DUSP5 mediated ERK signaling. Secondly, autophagy also links apoptosis and vascular calcifcation through AMPK/mTOR/ULK1, Wnt/β‑catenin and GAS6/AXL synthesis, as apoptotic cells become the nidus for calcium‑phosphate crystal deposition. The failure of mitophagy can activate Drp1, BNIP3, and NR4A1/DNA‑PKcs/p53 mediated intrinsic apoptotic pathways, which have been closely linked to the formation of vascular calcifcation. -

Thymocytes Trigger Self-Antigen-Controlling Pathways In
bioRxiv preprint doi: https://doi.org/10.1101/2020.11.29.399923; this version posted November 29, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Thymocytes trigger self-antigen-controlling pathways in immature medullary thymic epithelial stages Noëlla Lopes1, Nicolas Boucherit1, Jonathan Charaix1, Pierre Ferrier1, Matthieu Giraud2 and Magali Irla1,* 1Centre d’Immunologie de Marseille-Luminy, INSERM U1104, CNRS UMR7280, Aix- Marseille Université UM2, Marseille, 13288 cedex 09 France 2Inserm, Université de Nantes, Centre de Recherche en Transplantation et Immunologie, UMR 1064, ITUN, F-44000 Nantes, France *Correspondence: [email protected] 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.11.29.399923; this version posted November 29, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract Interactions of developing T cells with Aire+ medullary thymic epithelial cells expressing high levels of MHCII molecules (mTEChi) are critical for the induction of central tolerance. In turn, thymocytes regulate the cellularity of Aire+ mTEChi. However, it remains unknown whether thymocytes control Aire+ mTEChi-precursors that are contained in mTEClo cells or other mTEClo subsets that have recently been delineated or identified by single-cell transcriptomic analyses. Here, using three distinct transgenic mouse models, in which antigen-presentation between mTECs and CD4+ thymocytes is perturbed, we show by high-throughput RNA-seq that self- reactive CD4+ thymocytes induce in mTEClo the expression of tissue-restricted self- antigens, cytokines, chemokines and adhesion molecules important for T-cell development. -
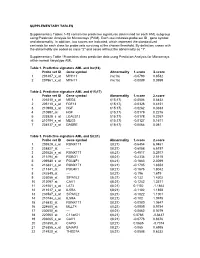
Supplementary Tables 1-18 Contain the Predictive Signatures Determined for Each AML Subgroup Using Prediction Analysis for Microarrays (PAM)
SUPPLEMENTARY TABLES Supplementary Tables 1-18 contain the predictive signatures determined for each AML subgroup using Prediction Analysis for Microarrays (PAM). Each row indicates probe set ID , gene symbol and abnormality. In addition, two scores are indicated, which represent the standardized centroids for each class for probe sets surviving at the chosen threshold. By definition, cases with the abnormality are coded as class "2" and cases without the abnormality as "1". Supplementary Table 19 contains class prediction data using Prediction Analysis for Microarrays within normal karyotype AML. Table 1. Predictive signature AML and inv(16) Probe set ID Gene symbol Abnormality 1.score 2.score 1 201497_x_at MYH11 inv(16) -0.0793 0.8532 2 207961_x_at MYH11 inv(16) -0.0089 0.0959 Table 2. Predictive signature AML and t(15;17) Probe set ID Gene symbol Abnormality 1.score 2.score 1 226210_s_at MEG3 t(15;17) -0.0506 0.6432 2 205110_s_at FGF13 t(15;17) -0.0326 0.4151 3 210998_s_at HGF t(15;17) -0.0262 0.3333 4 210997_at HGF t(15;17) -0.0179 0.2276 5 223828_s_at LGALS12 t(15;17) -0.0178 0.2267 6 210794_s_at MEG3 t(15;17) -0.0127 0.1611 7 204537_s_at GABRE t(15;17) -0.0064 0.081 Table 3. Predictive signature AML and t(8;21) Probe set ID Gene symbol Abnormality 1.score 2.score 1 205529_s_at RUNX1T1 t(8;21) -0.6454 6.9461 2 228827_at --- t(8;21) -0.6058 6.5197 3 205528_s_at RUNX1T1 t(8;21) -0.4917 5.2917 4 213194_at ROBO1 t(8;21) -0.2334 2.5115 5 206940_s_at POU4F1 t(8;21) -0.1883 2.0269 6 216831_s_at RUNX1T1 t(8;21) -0.1705 1.8353 7 211341_at -

BMP4 Induction of EKLF and GATA1 541 Were Obtained from Drs L
Development 129, 539-549 (2002) 539 Printed in Great Britain © The Company of Biologists Limited 2002 DEV4580 The BMP/BMPR/Smad pathway directs expression of the erythroid-specific EKLF and GATA1 transcription factors during embryoid body differentiation in serum-free media Carrie A. Adelman, Subrata Chattopadhyay and James J. Bieker* Department of Biochemistry and Molecular Biology, Mount Sinai School of Medicine, New York, NY 10029, USA *Author for correspondence (e-mail: [email protected]) Accepted 26 October 2001 SUMMARY Erythroid cell-specific gene regulation during terminal could be further stimulated by the inclusion of VEGF, SCF, differentiation is controlled by transcriptional regulators, erythropoietin and thyroid hormone. EBs were competent such as EKLF and GATA1, that themselves exhibit tissue- to respond to BMP4 only until day 4 of differentiation, restricted expression patterns. Their early expression, which coincides with the normal onset of EKLF expression. already in evidence within multipotential hematopoietic The direct involvement of the BMP/Smad pathway in this cell lines, has made it difficult to determine what induction process was further verified by showing that extracellular effectors and transduction mechanisms might erythroid expression of a dominant negative BMP1B be directing the onset of their own transcription during receptor or of the inhibitory Smad6 protein prevented embryogenesis. To circumvent this problem, we have taken induction of EKLF or GATA1 even in the presence of the novel approach of investigating whether the ability of serum. Although Smad1, Smad5 and Smad8 are all embryonic stem (ES) cells to mimic early developmental expressed in the EBs, BMP4 induction of EKLF and patterns of cellular expression during embryoid body (EB) GATA1 transcription is not immediate. -

Dynamic Regulation of Chromatin Accessibility by Pluripotency Transcription Factors
bioRxiv preprint doi: https://doi.org/10.1101/698571; this version posted July 11, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 Dynamic regulation of chromatin accessibility by pluripotency transcription factors 2 across the cell cycle 3 4 Elias T. Friman1, Cédric Deluz1, Antonio C.A. Meireles-Filho1, Subashika Govindan1, 5 Vincent Gardeux1, Bart Deplancke1, David M. Suter1 6 7 Corresponding author: [email protected] 8 9 Affiliations 10 1Institute of Bioengineering, School of Life Sciences, Ecole Polytechnique Fédérale de 11 Lausanne (EPFL), CH-1015 Lausanne, Switzerland 12 13 Abstract 14 The pioneer activity of transcription factors allows for opening of inaccessible regulatory 15 elements and has been extensively studied in the context of cellular differentiation and 16 reprogramming. In contrast, the function of pioneer activity in self-renewing cell divisions 17 and across the cell cycle is poorly understood. Here we assessed the interplay between 18 OCT4 and SOX2 in controlling chromatin accessibility of mouse embryonic stem cells. We 19 found that OCT4 and SOX2 operate in a largely independent manner even at co-occupied 20 sites, and that their cooperative binding is mostly mediated indirectly through regulation of 21 chromatin accessibility. Controlled protein degradation strategies revealed that the 22 uninterrupted presence of OCT4 is required for post-mitotic re-establishment and 23 interphase maintenance of chromatin accessibility, and that highly OCT4-bound enhancers 24 are particularly vulnerable to transient loss of OCT4 expression. Our study sheds light on 25 the constant pioneer activity required to maintain the dynamic pluripotency regulatory 26 landscape in an accessible state. -

BMC Molecular Biology Biomed Central
BMC Molecular Biology BioMed Central Research article Open Access Zinc-finger domains of the transcriptional repressor KLF15 bind multiple sites in rhodopsin and IRBP promoters including the CRS-1 and G-rich repressor elements Deborah C Otteson*1,2,4, Hong Lai2,5, Yuhui Liu2 and Donald J Zack1,2,3 Address: 1Guerrieri Center for Genetic Engineering and Molecular Ophthalmology at the Wilmer Eye Institute, Johns Hopkins University School of Medicine; 600 North Wolfe Street; Baltimore, MD 21287 USA, 2Department of Ophthalmology, Johns Hopkins University School of Medicine; 600 North Wolfe Street; Baltimore, MD 21287 USA, 3Departments of Neuroscience, and Molecular Biology and Genetics; Johns Hopkins University School of Medicine; 600 North Wolfe Street; Baltimore, MD 21287 USA, 4College of Optometry, University of Houston; Houston, TX 77204 USA and 5Department of Genetics, Stanford University School of Medicine; Stanford, CA 94305 USA Email: Deborah C Otteson* - [email protected]; Hong Lai - [email protected]; Yuhui Liu - [email protected]; Donald J Zack - [email protected] * Corresponding author Published: 17 June 2005 Received: 26 April 2005 Accepted: 17 June 2005 BMC Molecular Biology 2005, 6:15 doi:10.1186/1471-2199-6-15 This article is available from: http://www.biomedcentral.com/1471-2199/6/15 © 2005 Otteson et al; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Abstract Background: In the retina, many of the genes that encode components of the visual transduction cascade and retinoid recycling are exclusively expressed in photoreceptor cells and show highly stereotyped temporal and spatial expression patterns.