Studying BDNF Signalling Using Neurons Derived from Human Embryonic Stem Cells
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Screening and Identification of Key Biomarkers in Clear Cell Renal Cell Carcinoma Based on Bioinformatics Analysis
bioRxiv preprint doi: https://doi.org/10.1101/2020.12.21.423889; this version posted December 23, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Screening and identification of key biomarkers in clear cell renal cell carcinoma based on bioinformatics analysis Basavaraj Vastrad1, Chanabasayya Vastrad*2 , Iranna Kotturshetti 1. Department of Biochemistry, Basaveshwar College of Pharmacy, Gadag, Karnataka 582103, India. 2. Biostatistics and Bioinformatics, Chanabasava Nilaya, Bharthinagar, Dharwad 580001, Karanataka, India. 3. Department of Ayurveda, Rajiv Gandhi Education Society`s Ayurvedic Medical College, Ron, Karnataka 562209, India. * Chanabasayya Vastrad [email protected] Ph: +919480073398 Chanabasava Nilaya, Bharthinagar, Dharwad 580001 , Karanataka, India bioRxiv preprint doi: https://doi.org/10.1101/2020.12.21.423889; this version posted December 23, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract Clear cell renal cell carcinoma (ccRCC) is one of the most common types of malignancy of the urinary system. The pathogenesis and effective diagnosis of ccRCC have become popular topics for research in the previous decade. In the current study, an integrated bioinformatics analysis was performed to identify core genes associated in ccRCC. An expression dataset (GSE105261) was downloaded from the Gene Expression Omnibus database, and included 26 ccRCC and 9 normal kideny samples. Assessment of the microarray dataset led to the recognition of differentially expressed genes (DEGs), which was subsequently used for pathway and gene ontology (GO) enrichment analysis. -

Dissertation
Dissertation Submitted to the Combined Faculties for the Natural Sciences and for Mathematics of the Ruperto-Carola University of Heidelberg, Germany for the Degree of Doctor of Natural Sciences Presented by Ann-Cathrin Hofer (M.Sc.) Born in Heidelberg, Germany Oral Examination: 12th of September 2016 Regulatory T cells protect the neonatal liver and secure the hepatic circadian rhythm Referees 1st Referee: Prof. Dr. Peter Angel 2nd Referee: Dr. Markus Feuerer This dissertation was performed and written during the period from November 2012 to May 2016 in the German Cancer Research Center (DKFZ) under the supervision of Prof. Dr. Peter Angel and direct supervision of Dr. Markus Feuerer. The dissertation was submitted to the Combined Faculties for the Natural Sciences and for Mathematics of the Ruperto-Carola University of Heidelberg, Germany in June 2016. German Cancer Research Center (DKFZ) Immune Tolerance (D100) Im Neuenheimer Feld 280 69120 Heidelberg, Germany I II Confirmation Hereby, I confirm that I have written this thesis independently, using only the results of my investigation unless otherwise stated. Furthermore, I declare that I have not submitted this thesis for a degree to any other academic or similar institution. Parts of this dissertation have been submitted for publishing: Regulatory T cells protect the liver early in life and safeguard the hepatic circadian rhythm Ann-Cathrin Hofer, Thomas Hielscher, David M. Richards, Michael Delacher, Ulrike Träger, Sophia Föhr, Artyom Vlasov, Marvin Wäsch, Marieke Esser, Annette Kopp-Schneider, Achim Breiling, Frank Lyko, Ursula Klingmüller, Peter Angel, Jakub Abramson, Jeroen Krijgsveld & Markus Feuerer Parts of the experiments in this dissertation were performed in collaboration with other research groups as follows: CG methylation analysis with the 454 pyrosequencing technology: Division of Epigenetics, DKFZ, Heidelberg Dr. -
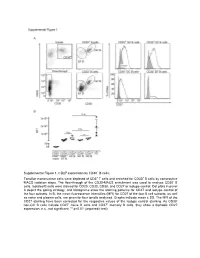
Supplemental Figure 1. CD27 Expression by CD30+ B Cells
Supplemental Figure 1. CD27 expression by CD30+ B cells. Tonsillar mononuclear cells were depleted of CD3+ T cells and enriched for CD30+ B cells by consecutive MACS isolation steps. The flow-through of the CD30-MACS enrichment was used to analyze CD30- B cells. Isolated B cells were stained for CD20, CD30, CD38, and CD27 or isotype control. Dot plots in panel A depict the gating strategy, and histograms show the staining patterns for CD27 and isotype control of the four subsets. In B, the mean fluorescence intensities (MFI) for CD27 of the four B cell subsets, as well as naive and plasma cells, are given for four tonsils analyzed. Graphs indicate mean ± SD. The MFI of the CD27 staining have been corrected for the respective values of the isotype control staining. As CD30- non-GC B cells include CD27- naïve B cells and CD27+ memory B cells, they show a biphasic CD27 expression. n.s., not significant; ** p<0.01 (unpaired t test). Supplemental Figure 2. Interleukin receptor expression by CD30+ B cells and relatedness of CD30+ EF B cells to CD21low B cells. mRNA and surface protein expression of IL2RB (A), IL21R (B) and CD21 (C) are shown for indicated B cell subsets of five tonsils (conventional (conv.) CD30- GC (germinal center) B cells (CD20highCD38+), CD30+ GC B cells, CD30- memory and CD30+ EF (extra follicular) B cells (CD20+CD38-/lowCD27+), and plasma cells (PC) (CD20+CD38high). mRNA data originate from Affymetrix genechip analyses (Tiacci et al., Blood, 2012; 120:4609-4620) and were analyzed using an unpaired t test. -

Resveratrol Is a Nuclear Receptor 4A1 (NR4A1) Ligand the Antagonizes NR4A1-Regulated Prooncogenic Pathways in Lung Cancers
Resveratrol is a Nuclear Receptor 4A1 (NR4A1) Ligand the Antagonizes NR4A1-Regulated Prooncogenic Pathways in Lung Cancers Lei Zhang1, Kumaravel Mohankumar1, Gregory Martin1, and Stephen Safe1 1Department of Veterinary Physiology and Pharmacology, Texas A&M University, College Station, TX, 77843 Abstract Methods A. C. Resveratrol (3,5,4’-trihydroxystilbene) is a polyphenolic phytochemical found in fruits, nuts, Ø Cell line: A549, H460 and H1299 non-small lung cancer cell lines were purchased and vegetables and there is evidence that this compound offers protection from several from American Type Culture Collection. human diseases including cancer. In cancer cell lines, resveratrol inhibits cell growth, survival, Ø Cell proliferation Assay: A549, H460, H1299 cells are seeded (2x104 per well) in migration/invasion and genes/pathways associated with these anticancer activities. Many of 96-well plates. A549 cells were maintained with DEME and 10% FBS. H460 and B. the same anticancer activities reported for resveratrol have previously been observed in this H1299 cells were maintained in RPMI 1640 and 10% FBS. Cells were allowed to laboratory using bis-indole derived nuclear receptor 4A1 (NR4A1) ligands that antagonize of attach for 24 hours. Then, the cells were treated at given concentrations with NR4A1-regulated pro-oncogenic pathways. Treatment of A549, H460, H1299 lung cancer cells appropriate media and 2.5% stripe FBS for 24, 48, and 72 hours. XTT cell viability with 50-125 µM resveratrol for 24, 48, 72 hours inhibited cell growth and IC50 values for kit that was purchased from Cell Signaling was applied to stain live cells after growth inhibition decreased with time. -

Figure S1. Gene Ontology Classification of Abeliophyllum Distichum Leaves Extract-Induced Degs
Figure S1. Gene ontology classification of Abeliophyllum distichum leaves extract-induced DEGs. The results are summarized in three main categories: Biological process, Cellular component and Molecular function. Figure S2. KEGG pathway enrichment analysis using Abeliophyllum distichum leaves extract-DEGs (A). Venn diagram analysis of DEGs involved in PI3K/Akt signaling pathway and Rap1 signaling pathway (B). Figure S3. The expression (A) and protein levels (B) of Akt3 in AL-treated SK-MEL2 cells. Values with different superscripted letters are significantly different (p < 0.05). Table S1. Abeliophyllum distichum leaves extract-induced DEGs. log2 Fold Gene name Gene description Change A2ML1 alpha-2-macroglobulin-like protein 1 isoform 2 [Homo sapiens] 3.45 A4GALT lactosylceramide 4-alpha-galactosyltransferase [Homo sapiens] −1.64 ABCB4 phosphatidylcholine translocator ABCB4 isoform A [Homo sapiens] −1.43 ABCB5 ATP-binding cassette sub-family B member 5 isoform 1 [Homo sapiens] −2.99 ABHD17C alpha/beta hydrolase domain-containing protein 17C [Homo sapiens] −1.62 ABLIM2 actin-binding LIM protein 2 isoform 1 [Homo sapiens] −2.53 ABTB2 ankyrin repeat and BTB/POZ domain-containing protein 2 [Homo sapiens] −1.48 ACACA acetyl-CoA carboxylase 1 isoform 1 [Homo sapiens] −1.76 ACACB acetyl-CoA carboxylase 2 precursor [Homo sapiens] −2.03 ACSM1 acyl-coenzyme A synthetase ACSM1, mitochondrial [Homo sapiens] −3.05 disintegrin and metalloproteinase domain-containing protein 19 preproprotein [Homo ADAM19 −1.65 sapiens] disintegrin and metalloproteinase -

Mediators in Human Disease and Autophagy
Chen et al. Cell Mol Biol Lett (2020) 25:48 https://doi.org/10.1186/s11658-020-00241-w Cellular & Molecular Biology Letters REVIEW LETTER Open Access The nuclear receptor 4A family members: mediators in human disease and autophagy Liqun Chen1,2*† , Fengtian Fan1,2†, Lingjuan Wu1,2 and Yiyi Zhao1,2 *Correspondence: [email protected] Abstract † Liqun Chen and Fengtian The Nuclear receptor 4A (NR4A) subfamily, which belongs to the nuclear receptor (NR) Fan are co-frst author. 1 College of Biological superfamily, has three members: NR4A1 (Nur77), NR4A2 (Nurr1) and NR4A3 (Nor1). Science and Engineering, They are gene regulators with broad involvement in various signaling pathways and Fuzhou University, human disease responses, including autophagy. Here, we provide a concise overview Fuzhou 350108, China Full list of author information of the current understanding of the role of the NR4A subfamily members in human is available at the end of the diseases and review the research into their regulation of cell autophagy. A deeper article understanding of these mechanisms has potential to improve drug development processes and disease therapy. Keywords: NR4A family, Autophagy, Human disease, Nur77, Nurr1, Nor1 Introduction Autophagy is an evolutionarily conserved catabolic process that attenuates cellular stress by digesting cytoplasmic contents and disposing of intracellular waste [1, 2]. Te process involves lysosomal enzymes degrading pathogens, longevity proteins, damaged orga- nelles and other components [3, 4]. Rapid adaptation to environmental changes to main- tain homeostasis is key to the health and survival of cells and organisms. By maintaining a balance between synthesis and degradation, autophagy can regulate homeostasis [5]. -

Molecular Insights Into NR4A2(Nurr1): an Emerging Target for Neuroprotective Therapy Against Neuroinflammation and Neuronal Cell Death
Molecular Neurobiology (2019) 56:5799–5814 https://doi.org/10.1007/s12035-019-1487-4 Molecular Insights into NR4A2(Nurr1): an Emerging Target for Neuroprotective Therapy Against Neuroinflammation and Neuronal Cell Death Md. Jakaria1 & Md. Ezazul Haque1 & Duk-Yeon Cho1 & Shofiul Azam1 & In-Su Kim1,2 & Dong-Kug Choi1,2 Received: 31 October 2018 /Accepted: 10 January 2019 /Published online: 25 January 2019 # Springer Science+Business Media, LLC, part of Springer Nature 2019 Abstract NR4A2 is a nuclear receptor and a transcription factor, with distinctive physiological features. In the cell nuclei of the central nervous system, it is widely expressed and identified as a crucial regulator of dopaminergic (DA) neuronal differentiation, survival, and maintenance. Importantly, it has regulated different genes crucial for dopaminergic signals, and its expression has been diminished in both aged and PD post-mortem brains and reduced in PD patients. In microglia and astrocytes, the expression of NR4A2 has been found where it can be capable of inhibiting the expression of proinflammatory mediators; hence, it protected inflammation-mediated DA neuronal death. In addition, NR4A2 plays neuroprotective role via regulating different signals. However, NR4A2 has been mainly focused on Parkinson’s research, but, in recent times, it has been studied in Alzheimer’s disease (AD), multiple sclerosis (MS), and stroke. Altered expression of NR4A2 is connected to AD progression, and activation of its may improve cognitive function. It is downregulated in peripheral blood mononuclear cells of MS patients; nonetheless, its role in MS has not been fully clear. miR-145-5p known as a putative regulator of NR4A2 and in a middle cerebral artery occlusion/reperfusion model, anti-miR-145-5p administration promoted neurological outcomes in rat. -

1 Transcriptomic Responses to Hypoxia in Endometrial and Decidual Stromal Cells 2 3 Kalle T
bioRxiv preprint doi: https://doi.org/10.1101/2019.12.21.885657; this version posted December 23, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 Transcriptomic responses to hypoxia in endometrial and decidual stromal cells 2 3 Kalle T. Rytkönen 1,2,3,4, Taija Heinosalo 1, Mehrad Mahmoudian 2,5, Xinghong Ma 3,4, Antti 4 Perheentupa 1,6, Laura L. Elo 2, Matti Poutanen 1 and Günter P. Wagner 3,4,7,8 5 6 1 Institute of Biomedicine, Research Centre for Integrative Physiology and Pharmacology, 7 University of Turku, Kiinamyllynkatu 10, 20014, Finland 8 2 Turku Bioscience Centre, University of Turku and Åbo Akademi University, Tykistökatu 6, 9 20520, Turku, Finland 10 3 Yale Systems Biology Institute, West Haven, Connecticut 06516, USA 11 4 Department of Ecology and Evolutionary Biology, Yale University, New Haven, CT 06511, 12 USA 13 5 Department of Future Technologies, University of Turku, FI-20014 Turku, Finland 14 6 Department of Obstetrics and Gynecology, Turku University Hospital, Kiinamyllynkatu 4-8, 15 20521, Turku, Finland. 16 7 Department of Obstetrics, Gynecology and Reproductive Sciences, Yale Medical School, New 17 Haven 06510, USA 18 8 Department of Obstetrics and Gynecology, Wayne State University, Detroit, MI- 48201, USA 19 20 Correspondence should be addresses to K T Rytkönen; Email: [email protected]. Address: Institute 21 of Biomedicine, Research Centre for Integrative Physiology and Pharmacology, University of 22 Turku, Kiinamyllynkatu 10, 20014, Finland / Turku Bioscience Centre, University of Turku and 23 Åbo Akademi University, Tykistökatu 6, 20520, Turku, Finland. -

A Bis-Indole-Derived NR4A1 Antagonist Induces PD-L1 Degradation and Enhances Anti-Tumor Immunity
Author Manuscript Published OnlineFirst on January 7, 2020; DOI: 10.1158/0008-5472.CAN-19-2314 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. A Bis-Indole-Derived NR4A1 Antagonist Induces PD-L1 Degradation and Enhances Anti-Tumor Immunity Keshav Karki1, Gus A. Wright2, Kumaravel Mohankumar1, Un-Ho Jin1, Xing-Han Zhang1 and Stephen Safe1 Affiliations: 1 Department of Veterinary Physiology and Pharmacology, Texas A&M University, College Station, TX, USA, 77843 2 Department of Veterinary Pathobiology, Texas A&M University, College Station, TX, USA, 77843 Running Title: NR4A1 antagonists as immunotherapy mimics Keywords: NR4A1, antagonists, mammary, tumors, immunotherapy mimics Corresponding Author: Stephen H. Safe, Ph. D., College of Veterinary Medicine & Biomedical Sciences, Department of Veterinary Physiology & Pharmacology, Texas A&M University, 4466 TAMU, College Station, TX 77843-4466 USA, Tel: 979-845-5988 / Fax: 979-862-4929, Email: [email protected] Disclosure of Conflicts of Interest: There are no conflicts of interests to declare. Word Count: 5004 Figures: 7 References: 46 1 Downloaded from cancerres.aacrjournals.org on October 6, 2021. © 2020 American Association for Cancer Research. Author Manuscript Published OnlineFirst on January 7, 2020; DOI: 10.1158/0008-5472.CAN-19-2314 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. ABSTRACT PD-L1 is expressed in tumor cells and its interaction with PD-1 plays an important role in evading immune surveillance; this can be overcome using PD-L1 or PD-1 immunotherapy antibodies. This study reports a novel approach for targeting PD- L1. -

Understanding the Role of the Transcription Factor Sp1 in Ovarian Cancer: from Theory to Practice
International Journal of Molecular Sciences Review Understanding the Role of the Transcription Factor Sp1 in Ovarian Cancer: from Theory to Practice Balachandar Vellingiri 1,*, Mahalaxmi Iyer 2, Mohana Devi Subramaniam 3, Kaavya Jayaramayya 2, Zothan Siama 4, Bupesh Giridharan 5, Arul Narayanasamy 6, Ahmed Abdal Dayem 7 and Ssang-Goo Cho 7,* 1 Human Molecular Cytogenetics and Stem Cell Laboratory, Department of Human Genetics and Molecular Biology, Bharathiar University, Coimbatore 641046, India 2 Department of Zoology, Avinashilingam Institute for Home Science and Higher Education for Women, Coimbatore 641043, India; [email protected] (M.I.); [email protected] (K.J.) 3 Department of Genetics and Molecular Biology, Vision Research Foundation, Sankara Nethralaya, Chennai 600006, India; [email protected] 4 Department of Zoology, School of Life-science, Mizoram University, Aizawl 796004, Mizoram, India; [email protected] 5 R&D Wing, Sree Balaji Medical College and Hospital (SBMCH), BIHER, Chromepet, Chennai 600044, Tamil Nadu, India; [email protected] 6 Disease Proteomics Laboratory, Department of Zoology, Bharathiar University, Coimbatore 641046, Tamil Nadu, India; [email protected] 7 Molecular & Cellular Reprogramming Center, Department of Stem Cell & Regenerative Biotechnology, Konkuk University, Seoul 05029, Korea; [email protected] * Correspondence: [email protected] or [email protected] (B.V.); [email protected] (S.-G.C.); Tel.: +45-24769924 (B.V.); +82-01-81743878 (S.-G.C.) Received: 15 January 2020; Accepted: 4 February 2020; Published: 9 February 2020 Abstract: Ovarian cancer (OC) is one of the deadliest cancers among women contributing to high risk of mortality, mainly owing to delayed detection. There is no specific biomarker for its detection in early stages. -

Autocrine IFN Signaling Inducing Profibrotic Fibroblast Responses by a Synthetic TLR3 Ligand Mitigates
Downloaded from http://www.jimmunol.org/ by guest on September 28, 2021 Inducing is online at: average * The Journal of Immunology published online 16 August 2013 from submission to initial decision 4 weeks from acceptance to publication http://www.jimmunol.org/content/early/2013/08/16/jimmun ol.1300376 A Synthetic TLR3 Ligand Mitigates Profibrotic Fibroblast Responses by Autocrine IFN Signaling Feng Fang, Kohtaro Ooka, Xiaoyong Sun, Ruchi Shah, Swati Bhattacharyya, Jun Wei and John Varga J Immunol Submit online. Every submission reviewed by practicing scientists ? is published twice each month by http://jimmunol.org/subscription Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://www.jimmunol.org/content/suppl/2013/08/20/jimmunol.130037 6.DC1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2013 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of September 28, 2021. Published August 16, 2013, doi:10.4049/jimmunol.1300376 The Journal of Immunology A Synthetic TLR3 Ligand Mitigates Profibrotic Fibroblast Responses by Inducing Autocrine IFN Signaling Feng Fang,* Kohtaro Ooka,* Xiaoyong Sun,† Ruchi Shah,* Swati Bhattacharyya,* Jun Wei,* and John Varga* Activation of TLR3 by exogenous microbial ligands or endogenous injury-associated ligands leads to production of type I IFN. -

TD GALVEZ CARVAJAL Laura.Pdf
UNIVERSIDAD DE MÁLAGA FACULTAD DE MEDICINA Departamento Medicina y Dermatología Tesis Doctoral IDENTIFICACIÓN DE UNA FIRMA DE EXPRESIÓN GÉNICA MEDIANTE EL SISTEMA DE ANÁLISIS NCOUNTER COMO MARCADOR PRONÓSTICO DE RECAÍDA EN PACIENTES CON TUMORES DE CÉLULAS GERMINALES NO SEMINOMA ESTADIO I Laura Gálvez Carvajal Málaga, 2020 AUTOR: Laura Gálvez Carvajal http://orcid.org/0000-0002-7952-598X EDITA: Publicaciones y Divulgación Científica. Universidad de Málaga Esta obra está bajo una licencia de Creative Commons Reconocimiento-NoComercial- SinObraDerivada 4.0 Internacional: http://creativecommons.org/licenses/by-nc-nd/4.0/legalcode Cualquier parte de esta obra se puede reproducir sin autorización pero con el reconocimiento y atribución de los autores. No se puede hacer uso comercial de la obra y no se puede alterar, transformar o hacer obras derivadas. Esta Tesis Doctoral está depositada en el Repositorio Institucional de la Universidad de Málaga (RIUMA): riuma.uma.es Revisión por Unicheck Cargado: 02/12/2020 | Verificado: 02/12/2020 Parte Matches Cita Referencia sustituida 0.7% Matches 22% Citas 0% Referencias Matches Web fuentes 153 1 www.laproteina.es 0.09% https://www.laproteina.es/tienda/3956-pure-bcaa-r811-300-gr.html 2 documentop.com 0.09% https://documentop.com/investigar-con-y-para-la-sociedad-uca_59fa65801723dd386241e125.html 3 mafiadoc.com 0.07% https://mafiadoc.com/utilizacion-de-material-didactico-con-recursos-de-ajedrez-para-la-_599c8d241723dd0f406ee02b.html 4 accessmedicina.mhmedical.com 0.04% https://accessmedicina.mhmedical.com/content.aspx?sectionid=114914497